Лечение суставов - артроз, артрит, остеохондроз и многое другое
Миопатия дюшенна что это такое
Миопатия Дюшенна: симптомы, механизм развития и прогноз
Мышечная дистрофия, или миопатия Дюшенна, — тяжелая наследственная патология, которая постоянно прогрессирует. Замедлить мышечное разрушение практически невозможно.
Связано это с врожденными изменениями. Впервые о миопатии Дюшенна заговорили в середине XIX века. Обнаружил эту патологию французский невролог. В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
Этот тип болезни сильно похож на миодистрофию Беккера, но в то же время отличается от него сложностью и внешними признаками.
Миодистрофия Дюшенна обнаруживает у 1 ребенка из 4000. Этот тип патологии относится к самым распространенным мышечным дистрофиям, относится к врожденным заболеваниям.
Этиология нарушения
Одному из генов в структуре генома человека присвоили имя невролога, в честь которого и было названо отклонение. На мышечную дистрофию Дюшенна могут влиять разные факторы:
- кровосмешение;
- предрасположенность генетического характера, например, при наличии миопатии Дюшенна у кого-либо из родни;
- неправильный синтез мышечных волокон, ускоренное распространение и замещение жировой прослойкой, соединительными волокнами;
- наследственные формы синдрома Дюшенна, чаще всего переходящие от матери;
- мутация генома при формировании во время беременности;
- аномалии в хромосомных структурах неясного происхождения;
- сильные нарушения в развитии дистрофина;
- патологические изменения биохимии в крови.
Характеристика наследственной патологии
Генетическая природа заболевания была сразу же доказана после обнаружения синдрома в 1868 году. Эта патология почти идентична с миодистрофией Беккера, то есть, обладает теми же генетическими предпосылками для формирования.
Однако миодистрофия Беккера отличается иными симптомами. Для болезни характерны следующие особенности:
- диагностируется у мальчиков до 5 лет;
- прогрессирует стремительно;
- у девочек никогда не обнаруживается;
- атрофия мышц обладает ступенчатым развитием – сначала страдает тазовый пояс;
- затем вовлекаются мышцы ног;
- после этого миопатия Дюшенна поражает мышцы спины, плеч;
- завершается прогрессирующая мышечная дистрофия Дюшенна поражением рук;
- специфический признак нарушения – деформация позвоночника, чаще встречающаяся в форме кифоза или лордоза;
- миодистрофия Дюшенна почти всегда сопровождается повреждениями грудины и стоп, они становятся неправильной формы, сильно меняют тело человека;
- при патологии, в отличие от миодистрофии Беккера, появляется повреждение левого сердечного желудочка, аритмия и кардиопатия;
- примерно у 30% пациентов развивается олигофрения.
Мышечная дистрофия Дюшенна никогда не протекает в легкой степени, всегда имеет крайне неблагоприятный прогноз. Развивается быстро, возможность ходить пациент утрачивает уже к 12 годам. При мышечной дистрофии Дюшенна смерть наступает из-за инфекции бронхов или легких, после остановки сердца.
Симптомы нарушения
Первые признаки миопатии Дюшенна встречаются уже в возрасте 1,5 лет. В редких случаях их не удается заметить до 5 лет. Проявляются признаки заболевания Дюшенна сначала в легкой степени. Их комбинация зависит от общего состояния здоровья:
- у ребенка возникает сильная неустойчивость, наблюдается неловкость в движении, он часто падает и очень медлителен;
- миопатия Дюшенна сопровождается тем, что во время ходьбы ребенок вихляет, постоянно спотыкается, в результате чего малыш боится вставать на ноги, возникает выраженная двигательная пассивность;
- со временем при мышечной дистрофии Дюшенна становится видна «утиная» походка с выпяченной вперед грудью и отведенными назад лопатками;
- если ребенок сидит или лежит, то принять стоячее положение при мышечной дистрофии Дюшенна становится сложно;
- при попытках принять стоячее положение, малыш словно встает на лесенку, поднимается вверх задом;
- возникает гипертрофия мышц, они заполняются жировой тканью;
- также миодистрофия Дюшенна захватывает работу сердца, в результате чего развиваются патологии и недостаточность;
- мышечная дистрофия Дюшенна часто сопровождается и другим признаком – появляются нарушения в биоптатах скелета;
- постепенно изменяется положение крупных суставов, начинается деформация стоп;
- миопатия Дюшенна в 100% случаев приводит к полной инвалидности пациента, ему требуется кресло;
- в 15 лет при мышечной дистрофии Дюшенна наступает глубокая инвалидизация, опасная остановкой сердца и хронические или постоянно рецидивирующие нарушения в легких.
На фоне мышечной дистрофии Дюшенна у маленького пациента развивается острая депрессия, которую дети с трудом переносят. Нередко причиной смертности при миодистрофии Беккера и Дюшенна становится суицид.
Диагностика заболевания
Мышечная дистрофия Дюшенна крайне тяжело поддается диагностики. Для этого привлекают комплекс методов. Первое, что нужно пройти при подозрении на миопатию Дюшенна, — это ЭКГ. Для подтверждения диагноза необходимо, чтобы анализ показал нарушения стенки левого желудочка.
Следующий этап – это определение уровня дистрофина, который не меняется в сторону обычной дистрофии. Также необходимо сдать кровь на биохимический анализ. Если есть миодистрофия Беккера или болезнь, названная в честь французского невролога, отмечается высокий уровень КФК.
Дополнительно нужно пройти ЭМГ, генодиагностику, а также биопсию мышц. Именно последний анализ позволяет установить болезнь с достаточно высокой точностью. Электромиография не уступает в эффективности с точки зрения постановки диагноза «мышечная дистрофия Дюшенна».
Тактика лечения заболевания
Чтобы лечение мышечной дистрофии Дюшенна было эффективным, нужно четко следовать намеченному плану после постановки диагноза. Излечению болезнь никогда не поддается полностью, но можно значительно облегчить жизнь пациента. Современная медицина способна замедлить миопатию Дюшенна следующими методами:
- Тактика при обнаружении болезни до 5 лет. Радикального лечения миодистрофии Дюшенна не требуется. Нужна генетическая консультация и постоянна поддержка родителей больного ребенка.
- Лечение миопатии Дюшенна до 8 лет. В этом случае нужна поддержка мышечных тканей. Врачи назначают глюкокортикостероиды для замедления прогресса болезни: «Преднизолон» или «Дефлазакорт».
- Терапия от 8 до 20 лет. В этом случае значительно ослабляются мышцы, мышечная дистрофия Дюшенна приводит ребенка к инвалидному креслу.
- Терапия от 20 лет. В этом случае препараты частично перестают действовать, прогрессируют заболевания дыхательных путей.
Миопатия Дюшенна требует постоянного приема некоторых групп витаминов (B, E), а также кальция, гормонов анаболиков, калия и некоторые виды аминокислот. Обязательно при мышечной дистрофии Дюшенна назначают инъекции АТФ, «Ретаболила», глютаминовой кислоты.
Важно! Поддержать здоровье при мышечной дистрофии Дюшенна можно и другими методами – ЛФК и электрофорезом.
ЛФК проходят небольшими курсами с обязательным участием терапевта. Также врачи рекомендуют делать массаж. Для электрофореза при миопатии Дюшеннанеобходимо использовать такие вещества, как липаза, хлорид кальция, «Прозерин».
В тяжелых случаях всё лечение проводят в домашних условиях, если есть медицинские возможности для организации сложной терапии специальными приборами.
Обязательное условие для лечения миопатии Дюшенна – постоянное наблюдение у кардиолога. Также необходимо составить грамотное меню. При заболевании нужно есть много овощей, приготовленных на пару, фруктов, растительных жиров и нежирного мяса. Запрещено употребление алкоголя, кофеина и крепкого чая.
Последствия и осложнения
В 100% случаев миопатия Дюшенна сопровождается тяжелыми последствиями для организма и сильно укорачивает жизнь. Пациент всегда умирает от осложнений заболевания – остановки сердца или легочной инфекции.
Если мышечную дистрофию Дюшенна удалось обнаружить в раннем возрасте, есть шанс, что человек доживет до 30 лет. Но только при условии адекватной терапии и комплексного подхода. Среди осложнений миопатии Дюшена нередко выделяют остеопороз, поражения позвоночника и суставов, а также патологии пищеварительной системы.
Мышечная дистрофия Дюшенна – тяжелое генетическое расстройство, терапия которого не способна оградить человека от одного исхода – смерти. В некоторых случаях пациентам удается прожить больше 20 лет после постановки диагноза. В других случаях младенцы умирают в течение первого года жизни.nevrology.net
Миопатия Дюшенна: причины, симптомы, лечение, как наследуется дистрофия, ее диагностика, фото
Мышечные дистрофии – это группа заболеваний не воспалительного характера, характеризующиеся прогрессивным течением без патологических изменений центральной и периферической нервной системы.
Что это такое?
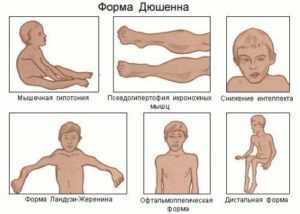 Фото 1. Признаки миопатии Дюшенна
Фото 1. Признаки миопатии Дюшенна Первые публикации о миопатиях датируются 1830 годом. В 1852 году Мерион сообщил о случае в семье с четырьмя сыновьями, у которых отмечались признаки двигательных расстройств, не связанных с поражением нервной системы. Он предположил, что заболевание наследуется от матери к ребенку.
Под миопатией или миодистрофией Дюшенна (также ее называют дистрофией или миопатией Дюшена-Беккера) понимают заболевание генетической природы, связанное с мутацией гена, кодирующего синтез белка дистрофина.
Дистрофин входит в состав мембран мышечных клеток, без него мышцы крайне уязвимы и подвергаются некрозу и дегенерации. Экспрессия, то есть реализация функции, происходит в гладкой и скелетной мышечной ткани, а также в миокарде.
Миопатия Дюшенна поражает одного из 3500 новорожденных детей. Несмотря на тот факт, что заболевание названо именем Дюшенна, одним из первых врачей, описавших данную патологию, был Говерс.
Гийом Дюшенн был французским неврологом, который использовал электростимуляцию в лечении неврологических расстройств. В 1868 году Дюшенн описал 13 пациентов с заболеванием, которое сопровождалось прогрессирующей мышечной слабостью, и назвал ее паралитической псевдогипертрофической мышечной дистрофией. Также он определил диагностические критерии миодистрофии, которые используются и сегодня.
Этиология, кто наследует и почему
По международной классификации болезней МКБ-10 мышечная дистрофия Дюшенна-Беккера имеет код G71.
Одна треть случаев заболевания связана со спонтанно возникшей мутацией, в то время как на остальные случаи патологии приходится наследование Х-хромосомы, несущей патологический ген.
Случаи гонадного мозаицизма связаны с 20% случаев миодистрофии Дюшенна. Средняя продолжительность жизни менее 20 лет. Повышение уровня фермента креатинфосфокиназы (КФК) обнаруживают у 2/3 женщин – носителей патологического гена. Большинство из них не имеют никаких проявлений заболевания. Миопатия почти всегда поражает мальчиков, поскольку заболевание сцеплено с Х-хромосомой, тип наследования – рецессивный. Локализация гена, кодирующего синтез дистрофина, находится в локусе Xq21. Синтез белка кодируется одним из самых больших известных генов. Он занимает около 2% ДНК Х-хромосомы.
Симптомы мышечной дистрофии
Первые симптомы появляются в возрасте от трех до семи лет. Обычно родители замечают раскачивающуюся походку и гиперлордоз. Существует несколько основных критериев, которые позволяют предположить миопатию Дюшенна. К ним относятся:
- Мышечная слабость, которая появляется неожиданно и начинается с нижних конечностей.
- Гиперлордоз – выраженный изгиб позвоночника кпереди, что особенно проявляется при ходьбе.
- Гипертрофия ослабленных мышц.
- Слабый ответ мышц на электрическую стимуляцию на более поздних стадиях заболевания.
С течением времени все указанные симптомы прогрессируют. Несмотря на поражение мышечных структур, нарушений функции мочевого пузыря и кишечника не отмечается. К двенадцати годам жизни большинство пациентов не могут ходить самостоятельно, и находится в инвалидном кресле. Миопатия Беккера очень сходна с дистрофией Дюшенна, так как поражается один и тот же локус Х-хромосомы, в результате чего страдает синтез дистрофина. Отличием миопатии Беккера является начало заболевания, как правило, после трех лет жизни или даже в подростковом возрасте.
Миодистрофия Дюшенна – патология, которая затрагивает не только скелетную мускулатуру. Дистрофин также содержится в миокарде, тканях мозга и гладкомышечной мускулатуре. Поздняя стадия заболевания ассоциируется с тяжелой сердечной недостаточностью и дыхательными нарушениями – главными причинами смерти пациентов.
Формы миопатии
В течении заболевания различают пять стадий. Первая стадия (доклинических проявлений) не характеризуется широким спектром симптомов, обычно у пациентов отмечается лишь повышение уровня КФК в сыворотке крови.
Вторая стадия (ранних признаков) включает следующие симптомы:
- Раскачивающаяся походка – впервые появляется от 2-х до 6-ти лет и часто является первым симптомом, который замечают родители.
- Прогрессирующая слабость мускулатуры нижних конечностей с дальнейшим присоединением слабости мышц шеи, плечевого пояса и рук.
- Наличие симптома Говерса – при попытке встать с пола, пациент упирается на колени и руки (рисунок 1). Появление симптома связано с выраженной слабостью мышц спины и конечностей.
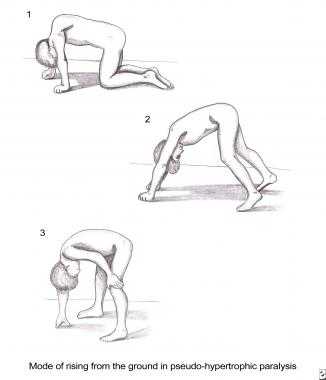 Рисунок 2. Симптом Говерса – пациент поднимается с пола, опираясь на колени и руки.
Рисунок 2. Симптом Говерса – пациент поднимается с пола, опираясь на колени и руки. Третья стадия (прогрессирующих симптомов) характеризуется появлением значительных сложностей во время ходьбы и развивается, когда возраст ребенка составляет около восьми лет. Пациенту становится сложно подниматься по ступеням, появляется одышка, сложно встать с пола. По утрам могут беспокоить головные боли, в ночное время наблюдается затрудненное дыхание. Четвертая и пятая стадии являются наиболее тяжелыми, так как пациент теряет возможность не только самостоятельно передвигаться, но и испытывает трудности с дыханием. При четвертой стадии пациент еще может удерживать осанку, однако делать это становится все труднее, развивается выраженный сколиоз.
Пятая стадия – терминальная. Пациент не может ходить и, как правило, находится в инвалидном кресле. Чаще всего, смерть наступает от дыхательной и сердечно-сосудистой недостаточности в возрасте до 20-ти лет.
Методы диагностики
Ребенок с миопатией Дюшенна-Беккера до 2-3-х лет жизни может ничем не отличаться от других детей. Тем не менее, существует ряд признаков, на которые стоит обратить внимание.
Важно! Во время проведения лабораторных исследований может быть повышен уровень печеночных ферментов: аланин- и аспартатаминотрансферазы, креатинкиназы и гамма-глутаматтрансферазы.
В ряде исследований миопатий было отмечено, что задержка речи и моторных навыков отмечалась чаще у детей с дефектом дистрофина. Уровень IQ может быть меньше на одно стандартное отклонение по сравнению со среднестатистическим значением в популяции.
У 30% детей с мутациями гена, кодирующего синтез дистрофина, отмечались трудности с обучением и приобретением новых навыков, обсессивно-компульсивные расстройства, синдром дефицита внимания, задержка умственного развития. Детям с дистрофиями Дюшенна и Беккера сложнее даются вербальные навыки.
Постановка диагноза «Миопатия Дюшенна» включает:
- Исследование уровня КФК, которая превышает референсные значения в 50-100 раз.
- Молекулярную диагностику – исследование гена, локализованного в локусе Xq21 и ответственного за синтез дистрофина, позволяет подтвердить или опровергнуть диагноз.
Если у пациента наблюдается сочетание высоких показателей КФК и мышечная слабость, это с высокой вероятностью позволяет заподозрить миопатию Дюшенна-Беккера.
Из инструментальных методов используют:
- обзорную рентгенографию грудной клетки. Исследование позволяет сделать заключение, насколько выражен сколиоз;
- электромиографию: метод применяют с целью дифференциальной диагностики со спинальной мышечной атрофией;
- электрокардиография – с целью выявления синусовых аритмий;
- ультразвуковое исследование сердца – нередко определяет малые размеры желудочков сердца и более длинную диастолу;
- холтеровское мониторирование определяет наличие пароксизмальных аритмий.
Если молекулярная диагностика не выявила мутации гена дистрофина, рекомендуется провести биопсию мышечной ткани. Характерные гистологические изменения приведены ниже:
- мышечные волокна с выраженным дегенеративным процессом и некрозом;
- пролиферация соединительной ткани;
- появление жировой ткани в значительном количестве.
Также проводится анализ белка дистрофина, выделенного из мышцы, с определением его молекулярной массы.
Лечение заболевания
На сегодняшний день лечения миодистрофии Дюшенна-Беккера не существует. Терапевтическая тактика основана на поддерживающем медикаментозном лечении (в основном, для сохранения сердечной функции), исследованиях в области генной инженерии и экспериментальном клеточном лечении.
При прогрессировании сколиоза и контрактур суставов возможно применение паллиативного хирургического вмешательства. По мере прогрессирования мышечной слабости, следует обеспечить пациенту максимальный комфорт.
Применение преднизолона активно обсуждается в профессиональных медицинских сообществах. Известно, что спустя один месяц от начала применения наблюдается незначительное улучшение общего состояния пациента и может сохраняться до трех лет без ухудшения. В то же время, при отказе от преднизолона начинается прогресс заболевания.
Последствия и осложнения
Главными осложнениями, с которыми сталкиваются пациенты, являются:
- Прогрессирующая мышечная слабость.
- Дилатационная кардиомиопатия.
- Дыхательные нарушения, связанные с дисфункцией диафрагмы.
- Контрактуры суставов.
- Сколиоз.
- Дисфагия.
- Запоры.
К осложнениям также относится остеопороз и высокий риск переломов. По некоторым данным возможно использование препаратов кальция и витамина Д для увеличения плотности костной ткани.
Прогноз на выздоровление и жизнь
К сожалению, прогноз неблагоприятный, так как смертность при данной патологии составляет 100%. Если в семейной истории встречается близкая родственница, которая является носителем патологического гена, у детей мужского пола выше риск развития кардиомиопатии с прогрессирующей сердечной недостаточностью в возрасте от 20 до 40 лет. Проводятся исследования, связанные с применением стволовых клеток для лечения миодистрофий.
Что нужно запомнить?
- Миопатия Дюшенна-Беккера – прогрессирующее заболевание, связанное с аномальным синтезом белка дистрофина, принимающего непосредственное участие в мышечных сокращениях. Синтез дефектного белка приводит к дегенеративным и некротическим процессам в мышцах.
- Ранними признаками болезни считаются появление мышечной слабости в нижних конечностях, раскачивающейся, нетипичной походки. К лабораторным признакам относится многократное повышение уровня КФК.
- Этиология заболевания – генетическая, связанная с мутацией гена в локусе Тип наследования-рецессивный, мутация может передаваться от матери, чья Х-хромосома несет патологический ген, а может возникать спонтанно.
- Существует пять стадий миопатии. Четвертая и пятая стадии являются терминальными.
- К ранним методам диагностики относится определение уровня КФК, который может многократно превышать норму и являться ранним маркером наследственной миодистрофии Дюшенна-Беккера задолго до появления мышечной слабости и других заметных симптомов. К более точным методам относится молекулярная диагностика с исследованием патологического гена.
- Специфическое лечение не разработано. Мероприятия по уходу за пациентом сводятся к поддержанию жизнеобеспечения.
- Основными осложнениями являются утрата способности ходить, тяжелые дыхательные расстройства и сердечно-сосудистая недостаточность.
- Прогноз неблагоприятный, летальность составляет 100%.
Литература
- Bushby K, Straub V. Nonmolecular treatment for muscular dystrophies. Curr Opin Neurol. 2005 Oct. 18(5):511-8.
- Moxley RT 3rd, Ashwal S, Pandya S, Connolly A, Florence J, Mathews K, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005 Jan 11. 64(1):13-20.
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991 Sep 20. 66(6):1121-31.
- Ozawa E, Noguchi S, Mizuno Y, et al. From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve. 1998 Apr. 21(4):421-38.
- Darke J, Bushby K, Le Couteur A, McConachie H. Survey of behaviour problems in children with neuromuscular diseases. Eur J Paediatr Neurol. 2006 May. 10(3):129-34.
- Mendell JR, Shilling C, Leslie ND et al. Evidence based path to newborn screening for Duchenne Muscular Dystrophy. Ann Neurology. 2012. 71:304–313.
- Rodino-Klapac LR, Chicoine LG, Kaspar BK, Mendell JR. Gene therapy for duchenne muscular dystrophy: expectations and challenges. Arch Neurol. 2007 Sep. 64(9):1236-41.
- Merlini L, Gennari M, Malaspina E et al. Early corticosteroid treatment in 4 duchenne muscular dystrophy patients: 14-year follow-up. Muscle Nerve. 2012 Jun. 45(6):796-802.
- American Academy of Pediatrics Section on Cardiology and Cardiac Surgery. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005 Dec. 116(6):1569-73.
- Colan SD. Evolving therapeutic strategies for dystrophinopathies: potential for conflict between cardiac and skeletal needs. Circulation. 2005 Nov 1. 112(18):2756-8.
doktor-ok.com
Миопатия Дюшена: фото, причины, симптомы, диагностика, лечение и профилактика
Одной из самых грозных первичных мышечных дистрофий, которая начинается в раннем детстве и приводит к летальному исходу до достижения больным 25-летнего возраста, является миопатия Дюшена (полное название – прогрессирующая мышечная дистрофия Дюшена).
Краткая характеристика миопатии Дюшена
Заболевание было впервые описано в 1868 году Дюшеном (Duchenne) и является генетическим. Причем миопатия Дюшена имеет общую, генетически единую форму с миопатией Беккера, однако отличается целым рядом клинических признаков.
Встречается болезнь у одного из 3-3,5 тысяч новорожденных мальчиков и обнаруживается в возрасте 1,5-3 лет и быстро прогрессирует.
Обычно больные не доживают даже до 30-летнего возраста (по некоторым данным, и вовсе многие умирают в возрасте 20-22 лет).
Распространению процесса гипотрофии мышц свойственен восходящий характер:
- Сначала в процесс вовлекаются мышцы тазового пояса, а также мышцы проксимальных отделов ног (нижних конечностей),
- Затем страдают мышцы спины и плечевого пояса,
- После этого очередь доходит до проксимальных отделов рук (верхних конечностей).
Уже в самом начале заболевания угасают или значительно снижаются коленные рефлексы, при этом сухожильные рефлексы рук и Ахиллов рефлекс сохраняются ещё очень длительное время.
Другие признаки этого заболевания:
- Прогрессирование кифоза, лордоза или других вторичных деформаций позвоночника,
- Искривление грудной клетки (она становится килевидной или седловидной), стоп,
- Развиваются ретракции сухожилий на фоне контрактур в суставах.
- Очень часто при миопатии Дюшена наблюдаются проблемы с сердцем, а именно кардиомиопатия, симптомами которой становятся аритмия и гипертрофия левого желудочка.
- Хотя обычно при миопатиях интеллект не страдает, в этом случае у 25-30% пациентов наблюдается олигофрения (а именно, в степени дебильности). У остальных больных интеллект сохранен.
Прогноз заболевания неблагоприятный – миодистрофия Дюшена быстро прогрессирует, больные теряют способность к самостоятельной ходьбе после 10-12 лет, а умирают в молодом возрасте по причине интеркуррентных инфекций (дыхательной недостаточности) или от сердечной недостаточности.
Фото миопатии Дюшена: 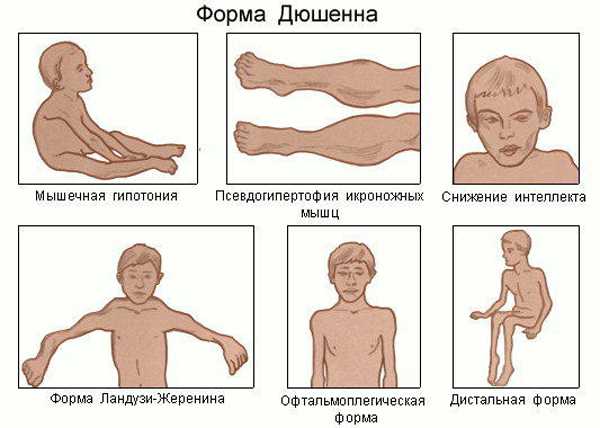
Причины
Миопатия Дюшена является наследственным заболеванием, причем носителями его гена являются представительницы женского пола. Наследуется эта миопатия по рецессивному типу, сцепленному с Х-хромосомой. Более того, около трети всех случаев выявления миопатии Дюшена вызваны новыми мутациями генов. Болеют исключительно мальчики.
Из всего многообразия миопия эта является наиболее злокачественной и быстро прогрессирующей.
Хотя миопатия Дюшена является наследственным заболеванием, в 30% случаев причиной является генная мутация.
Симптомы заболевания
Заболевание начинает проявляться в возрасте 1,5-5 лет, его первыми признаками являются:
- Неустойчивость, двигательная неловкость.
- Постоянные спотыкания и падения во время ходьбы, что развивает у ребенка страх перед ходьбой, чем обусловлена двигательная пассивность.
- Ребенку сложно подниматься по лестнице, а походка становится вразвалочку, «утиной».
- Также затруднительным становится подъем из положения лежа или сидя – ребенок прибегает к так называемым «приемам Гроверса» — это «взбирание по самому себе» и «взбирание лесенкой».
- Яркий признак миопатии Дюшена – мнимая гипертрофия мышц, особенно икроножных: на самом деле происходит не развитие мышц, а их перерождение в жировую и соединительную ткани.
- Как уже упоминалось, одним из симптомов миопатии Дюшена является поражение сердца, причиной которого, по мнению зарубежных исследователей, становится недостаток в кардиомиоцитах дистрофина.
- Наблюдаются признаки миопатии в биоптатах мышц скелета.
- Со временем, по мере прогрессирования мышечной дистрофии развиваются контрактуры крупных суставов, наблюдается эквиноварусная деформация стопы.
- Ближе к 10-12 годам ребенок уже не может передвигаться самостоятельно и вынужден пользоваться инвалидным креслом.
- К 15 годам развивается глубокая инвалидизация больного.
Знаете ли вы, какие бывают симптомы серотонинового синдрома? Причины и последствия.
Всё о принципах лечения миофасциального болевого синдрома можно найти здесь.
Диагностика
Диагностика миодистрофии Дюшена ставится на основании следующих результатов осмотра и анализов:
- На ЭКГ выявляется поражение миокарда латеральной и задне-нижней стенок левого желудочка, что определяется по следующим показателям: высокий зубец наблюдается в отведении V6; глубокий зубец Q наблюдается в отведениях V6, aVF, 2 и 3.
- Также исследуется содержание дистрофина в мышечной ткани (при этом заболевании дистрофия не выявляется).
- В ходе биохимических исследований в плазме крови определяется активность КФК (фермента креатинфорсфокиназы), которая обычно существенно повышена (в том числе у носительниц гена). Иногда для уточнения источника исследуются изоферменты КФК.
- Проводится также генодиагностика.
- Фибрилляции на ЭМГ сообщают о некрозе мышечных волокон.
- Биопсия мышц является одним из основных методов диагностики миопатии Дюшена, причем выбирается умеренно пораженная мышца, поскольку очень ослабленная и существенно пострадавшая мышца окажется неинформативной.
Наиболее достоверными являются анализы на активность в сыворотке мышечных ферментов, биопсия мышц и ЭМГ (электромиография).
Лечение миопатии Дюшена
При таком тяжелом и быстро прогрессирующем заболевании лечение малоэффективно, обычно в комплексной поддерживающей терапии применяются следующие препараты:
- Группа препаратов, улучшающих обмен веществ в организме: витамины группы В, Е, аминокислоты, препараты кальция, анаболические гормоны, калия оротат.
- Применяется лечение прозерином, галантамином, оксазилом.
- Лечение проводится курсами и, предпочтительнее, в стационаре: ЛФК (особенно замедляющая образование контрактур), так же, как и пассивное растяжение больных мышц, массаж, электрофорез прозерина, лидазы, кальция хлорида, ванны, индуктотермия. Курс лечения повторяется каждые 6-8 недель, а неподвижных детей предпочтительнее лечить на дому.
- Последние годы популярным стало лечение глюкокортикоидами (по схеме через день), которое продлевает жизнь ещё на несколько лет.
Больному миопатией Дюшена нужно обязательно наблюдаться у врача-кардиолога.
Отдельно стоит отметить важность полноценного питания, в котором обязательно должны присутствовать растительные жиры и белки животного происхождения. Стоит избегать крепкого чая, кофе, алкоголя, пряностей, сахара, капусты и картофеля. В рацион должны входить свежие или приготовленные щадящим способом овощи, фрукты, кисло-молочные продукты, овсяная крупа, яйца, мед, морковь, орехи.
Не секрет, что нервными тиками страдают как взрослые люди, так и дети. Причины нервного тика глаза, диагностика и лечение.
Симптомы алкогольной энцефалопатии хорошо освещены в этой статье.
Лечение нейропатической боли у взрослых зачастую представляет собой длительный процесс и требует комплексного подхода. Подробную информацию об этом заболевании можно получить, перейдя по этой ссылке: http://gidmed.com/bolezni-nevrologii/nevralgija/nejropaticheskaya-bol.html.
Профилактика
Профилактика заболевания затруднена ввиду генетических причин его возникновения, вот почему особо важную роль играет генетическое консультирование тех семей, в которых выявлена отягощенная наследственность.
Новейшие методы и разработки молекулярной генетики помогают достоверно определить природу мутации гена, «вычислить» прогноз его заболевания, но самое главное – такие методы позволяют провести перинатальное обследование в случае повторной беременности.
Оцените статью: (3 оценок, среднее: 5,00 из 5)gidmed.com
Миопатия Дюшенна: причины, симптомы и лечение миодистрофии

Поделиться
Поделиться
ПоделитьсяПоделиться
Мышечная дистрофия Дюшенна (МДД) или миопатия Дюшена — это врожденное заболевание, которое вызывает прогрессирующую слабость мышц. Это заболевание проявляется в детстве — родители ребенка могут заметить, что ему трудно стоять, бегать или карабкаться на лесенки на детских площадках. Болеют этим видом миопатии преимущественно мальчики, но девочки могут быть носителями так называемого гена Дюшена (известны крайне редкие случаи, когда от этого заболевания страдали и девочки). Пациентам с таким диагнозом приходится часто обследоваться у врача, а с определенного возраста (в среднем — с девяти лет) у них возрастает потребность в средствах для облегчения признаков этого нарушения.
Что такое миопатия Дюшена
Это серьезное заболевание поражает, главным образом, мышцы в области туловища, бедер и плеч. При этом пациенты, как правило, могут свободно использовать руки и пальцы, но у них возникают проблемы с ходьбой, бегом, и так далее. Мышечная слабость прогрессирует постепенно. Обычно она проявляется в раннем детстве, но поначалу симптомы миопатии Дюшена выражены очень слабо. С возрастом они становятся все более выраженными, и приводят к резкому снижению качества жизни.
Миопатия Дюшена диагностируется приблизительно у одного из 3500 мальчиков.
В мышечной ткани содержится дистрофин — белок, необходимый для нормальной работы мышц. У людей с миопатией Дюшена этого вещества слишком мало. Со временем это приводит к повреждению мышечных волокон и ослаблению мышц. Причиной этого является особый ген, который передается от родителей к детям, либо генные мутации, произошедшие в период внутриутробного развития.
Для каждого сына женщины, которая является носителем гена Дюшена, вероятность развития миопатии Дюшена составляет ровно 50%. Дочери такой женщины станут носителями этого гена с такой же вероятностью.
Если у ребенка миопатия Дюшена, значит ли это, что у кого-то из членов семьи есть ген Дюшена? Не обязательно. Приблизительно в половине случаев заболевшие миопатией этого типа не получают дефектный ген от одного из родителей. В клетках плода еще во время беременности происходят мутации, результатом которых и становится миопатия. Это может произойти из-за «ошибки», которая случилась при копировании родительских генов в клетки, которые должны будут образовать организм ребенка. Почему это происходит, в настоящее время неизвестно.
Точно узнать, есть ли у кого-то из членов вашей семьи ген Дюшена, можно, только при помощи генетического консультирования.
Каковы симптомы?
Обычно первые симптомы миопатии Дюшена появляются в возрасте 1-3 лет. Родители могут заметить следующие признаки миопатии Дюшена:
- Ребенку трудно ходить, бегать, прыгать, подниматься по лестницам. Походка ребенка может отличаться от походки его ровесников — он ходит вразвалочку, и менее уверенно, чем остальные. Иногда дети с миопатией Дюшена начинают ходить позже остальных, однако и совершенно здоровые малыши иногда делают первые шаги несколько позже сверстников;
- В более старшем возрасте ребенок может опираться на руки, чтобы встать;
- У ребенка могут наблюдаться проблемы с обучением — как правило, не очень серьезные.
Иногда первым признаком миопатии Дюшена является замедленное развитие речи.
Как диагностируется
В первую очередь врачи, как правило, просто наблюдают за ребенком, в особенности за тем, как он ходит, бегает и встает с пола. Если основания подозревать миопатию Дюшена, будет назначен анализ крови на креатинкиназу — это фермент, уровень которого у людей с этим нарушением всегда очень высок (в 10-100 раз выше нормы). Если уровень креатинкеназы у ребенка в норме, миопатию Дюшена исключают и начинают искать другие причины появившихся у малыша симптомов.
Следующим этапом диагностики миопатии Дюшена является биопсия мышечной ткани и/или генетическое тестирование.
В ходе биопсии врач берет небольшой фрагмент мышечной ткани для дальнейших анализов; процедуру проводят под общей анестезией. Образец ткани изучают под микроскопом при помощи особых техник, чтобы оценить состояние мышечных волокон и количество дистрофина.
Для проведения генетического тестирования необходимо необходимое количество крови пациента. С помощью этого метода выявляют гены, которые отвечают за развитие миопатии Дюшена. В большинстве случаев этот способ позволяет точно диагностировать данное заболевание.
В настоящее время вылечить эту болезнь невозможно. Однако существует ряд способов, которые могут облегчить состояние пациента на разных стадиях развития миопатии и, возможно, несколько замедлить прогрессирование заболевание.
Ниже описаны способы лечения для определенных возрастных групп, но нередко при лечении одного пациента приходится сочетать сразу несколько методов терапии.
Пациенты дошкольного возраста
Обычно в этом возрасте детям с миопатией Дюшена еще не нужно лечение. Родителям могут предложить:
- Подробную информацию о миопатии Дюшена. Врач проведет с родителями беседу и подробно расскажет о том, как эта болезнь будет влиять на состояние ребенка, и какова ожидаемая продолжительность жизни пациента (большинство людей с таким диагнозом доживают лишь до 20-25 лет). При желании родители могут обратиться к другим специалистам (например, к психологу), а также в группы поддержки;
- Рекомендации относительно допустимых физических нагрузок для ребенка;
- Генетическую консультацию для членов семьи. Многие родители желают узнать, являются ли они носителями гена Дюшена. Это особенно важно для тех, кто в будущем планирует снова родить ребенка.
Уже в дошкольном возрасте ребенка начинают регулярно обследовать, чтобы, когда это будет необходимо, можно было вовремя начать лечение.
Пациенты в возрасте 5-8 лет
Детям такого возраста может потребоваться поддержка для мышц ног. Например, им могут рекомендовать надевать на ночь шины для лодыжки или более длинные шины, для голени.
При помощи кортикостероидов можно замедлить развитие миопатии и в течение некоторого времени сохранять мышцы достаточно сильными. Пациенты постоянно или курсами принимают такие препараты, как преднизолон или дефлазакорт. Поскольку кортикостероиды могут вызывать серьезные побочные эффекты, ребенок обязательно должен наблюдаться у врача.
Пациенты от 8 лет до позднего переходного возраста
Через какое-то время после достижения ребенком возраста восьми лет его мышцы начинают заметно слабеть. Ходить со временем становится все труднее пока, наконец, ребенку не придется начать передвигаться на инвалидном кресле. Возраст, в каком это происходит, варьируется от пациента к пациенту. Часто это случается между 9 и 11 годами, но дети, которые достаточно рано начали принимать кортикостероиды, иногда могут продолжать ходить несколько дольше.
Вскоре после того, как ребенку для передвижения становится необходимым инвалидное кресло, у него начинают развиваться и другие осложнения, поэтому ему могут потребоваться более частые обследования. Все осложнения необходимо начинать лечить как можно раньше.
Кроме этого, родителям нужно позаботиться о практической стороне жизни ребенка — на первых порах помогать ему передвигаться в кресле, а также по возможности приспособить свой дом под его нужды.
Пациенты от позднего переходного возраста до 20+ лет
В этом возрасте мышечная слабость вызывает все больше проблем, и пациенту все чаще требуется помощь других людей. Увеличивается вероятность развития тяжелых осложнений, таких как легочные инфекции.
Как говорилось выше, миопатия Дюшена — тяжелое заболевание, которое значительно сокращает жизнь человека. Со временем мышечная слабость вызывает все более серьезные проблемы с дыхательной системой и работой сердца. В прошлом большинство пациентов с миопатией Дюшена доживали лишь до 20-23 лет. Сегодня все больше людей с этим диагнозом доживают до 27 лет, а иногда и до более старшего возраста. Отметим, что продолжительность жизни зависит от многих факторов, таких как сопутствующие заболевания, доступность качественной медицинской помощи, и так далее. Со временем ожидаемая продолжительность жизни при миопатии Дюшена может еще больше увеличиться.
Наиболее распространенной причиной смерти больных являются осложнения, связанные с респираторной системой, например, тяжелые инфекции дыхательных путей.
Осложнения при миопатии Дюшена и их лечение
Остеопороз. У больных миопатией Дюшена может развиваться остеопороз — заболевание, которое приводит к уменьшению плотности костной ткани. Основными причинами этого являются вынужденная малоподвижность и прием кортикостероидов. Очень важно как можно дольше предотвратить развитие этой болезни. Для этого необходимо получать достаточное количество витамина D и кальция — они требуются для того, чтоб кости оставались крепкими. Эти вещества можно получить как из пищи, так и из витаминных добавок.
Пациентам, у которых уже развился остеопороз, назначают лекарственные препараты, например, бисфосфонаты.
Осложнения, затрагивающие суставы и позвоночник
Мышечная слабость может стать причиной того, что суставы пациента со временем станут менее подвижными. В таких случаях пациентам могут рекомендовать носить шины, а иногда может потребоваться хирургическое вмешательство.
Сколиоз, или искривление позвоночника, также может стать результатом прогрессирующей мышечной слабости. Обычно он развивается уже после того, как пациент начинает передвигаться в инвалидном кресле. Чаще всего для лечения сколиоза пациенты носят специальные корсеты. Хирургические операции при этом нарушении назначают лишь в редких случаях.
Осложнения, связанные с питанием и пищеварением
У детей с миопатией Дюшена часто бывает избыточный вес, особенно если они принимают кортикостероиды. У подростков и взрослых людей с этим диагнозом, напротив, может быть дефицит массы тела из-за постепенного разрушения мышечных волокон. Чтобы удержать вес в пределах нормы, пациентам следует регулярно консультироваться с диетологами, и выполнять их рекомендации.
Запор часто становится результатом малоподвижного образа жизни. Для лечения запоров назначается прием слабительных препаратов, а для их профилактики рекомендуется употреблять в пищу больше продуктов, богатых диетической клетчаткой.
На поздних стадиях развития миопатии Дюшена — в возрасте старше 18-20 лет — у многих пациентов появляются проблемы с жеванием и глотанием пищи. В наиболее тяжелых случаях может быть необходима гастростомия — операция, в ходе которой в полость желудка вводят специальную трубку, через которую будет осуществляться кормление.
Осложнения, связанные с дыхательной системой
В подростковом возрасте дыхательные мышцы пациентов начинают ослабевать, из-за чего дыхание становится поверхностным, а кашлевой механизм становится менее эффективным. Это может привести к различным респираторным инфекциям, поскольку слизь и бактерии выводятся из дыхательных путей не так легко, как у здоровых людей. Важно лечить такие инфекции своевременно — обращаться к врачу при появлении первых симптомов, и принимать все назначенные лекарства. Для профилактики некоторых инфекционных заболеваний пациенту могут сделать прививки.
По мере того, как дыхательные мышцы слабеют, концентрация кислорода в крови снижается, особенно во время сна. Поскольку это происходит постепенно, признаки этого поначалу могут быть незаметны. Наиболее распространенными симптомами понижения уровня кислорода в крови являются головные боли по утрам, частые пробуждения по ночам, слабость, раздражительность, очень живые, насыщенные сновидения.
Своевременное обращение к врачу позволяет облегчить многие из этих симптомов, и заметно улучшить качество жизни.
Осложнения, связанные с работой сердца
У подростков и взрослых с миопатией Дюшена может развиться кардиомиопатия — нарушение, для которого характерна слабость сердечной мышцы. Признаками кардиомиопатии могут быть повышенная утомляемость, одышка, отеки ног, нерегулярное сердцебиение. Для лечения кардиомиопатии назначается медикаментозная терапия. Чем раньше ее начать, тем более эффективной она будет.
Источник http://www.womenclub.ru/
Поделиться
Поделиться
ПоделитьсяПоделиться
Что такое мышечная дистрофия Дюшенна?
Мышечная дистрофия Дюшенна – это мышечная атрофия, вызванная недостатком белка под названием дистрофин. Обычно этой болезни подвержены только мальчики. Около 100 мальчиков с мышечной дистрофией Дюшенна рождаются в Соединенном Королевстве каждый год, и на данный момент на территории Королевстве зарегистрировано 2500 мальчиков и молодых людей с мышечной дистрофией Дюшенна. Возможен риск, что из 3500-5000 младенцев мужского рода один будет с мышечной дистрофией Дюшенна.
Мышечная дистрофия Дюшенна – это серьезное заболевание, которое вызывает прогрессирующее ослабление мышц. Из-за нехватки дистрофина мышечные волокна ослабевают и уступают место волокнистой или жировой ткани, вызывая постепенное разрушение мышц.
Каковы причины мышечной дистрофии Дюшенна?
Мышечная дистрофия Дюшенна – это генетическое заболевание, вызванное отклонением или мутацией в генетическом коде (ДНК). При мышечной дистрофии Дюшенна мутация возникает в гене, который называется дистрофин и который расположен в Х-хромосоме или половой хромосоме (у девочек две Х-хромосомы, у мальчиков – только одна). В половине случаев заболевание передается от матери, являющейся «носителем», но оно также может стать результатом новой мутации в генах ребенка.
Если мутация происходит в организме женщины, то она называется «носителем». Обычно носители женского пола не подвержены этому заболеванию, поскольку у них есть вторая Х-хромосома, где дистрофин может вырабатываться. У небольшого количества женщин есть некоторая степень мышечной слабости, их называют «явными носителями».
У каждого сына носителя есть риск 50%, что он будет подвержен этому заболеванию, а у каждой дочери – риск 50%, что она – носитель.
Консультация генетиков и обследование остальных членов семьи на риск быть носителями должны быть проведены как можно скорее, как только мальчику будет поставлен диагноз мышечной дистрофии Дюшенна. Вы можете обратиться к Вашему врачу-консультанту или семейному врачу.
Как распознают мышечную дистрофию Дюшенна?
У большинства мальчиков с мышечной дистрофией Дюшенна это заболевание не обнаруживают, пока не появятся соответствующие симптомы, за исключением тех случаев, когда в семье есть человек с таким заболеванием. Первые признаки мышечной дистрофией Дюшенна обычно появляются в возрасте от одного до трех лет и, как правило, связаны с нарушением функционирования мышц. Мальчики могут начать ходить позже, чем их сверстники, падать чаще или испытывать трудности с бегом, прыжками или подъемом. У них могут быть увеличены икроножные мышцы.
У некоторых мальчиков с мышечной дистрофией Дюшенна замедленная речь, что может стать первым признаком этого заболевания. При сдаче анализа крови обнаруживается высокое содержание белка под названием креатинкиназа (КК). КК, как правило, появляется в мышце, но при их повреждении, как в случае с мышечной дистрофией Дюшенна, он попадает в кровь. Ферменты печени (аминотрансферазы, ALT и AST) также часто находятся на высоком уровне, как следствие повреждения мышц, а не нарушений печени.
Мышечная дистрофия Дюшенна должна быть подтверждена генетическим анализом крови. Разные типы генетических анализов предоставляют специфические и подробные данные о мутации ДНК.
Генетическое подтверждение заболевание имеет решающее значение. Это позволяет семьям проводить пренатальную диагностику будущих беременностей, а также обследовать остальных членов семьи на случай, если они являются носителями мутации в гене дистрофина. Более того, генетическая диагностика поможет определить, может ли мальчик претендовать на участие в клиническом исследовании, которое начинается или запланировано.
Ваш врач также может посоветовать пункционную биопсию мышц, когда берут небольшой образец мышцы для анализа. Такие исследования могут предоставить информацию о том, какое количество белка дистрофина присутствует в клетках мышцы, а также в некоторых случаях помочь отличить мышечную дистрофию Дюшенна от слабой формы заболевания, известной как мышечная дистрофия Беккера. Однако, как правило, достаточно клинических признаков и генетического анализа, чтобы различить эти две формы, без необходимости мышечной биопсии.
Существует ли лечение или лекарство?
Пока не было изобретено лекарства для этого заболевания, но есть обнадеживающие исследования в этой области. Подход с привлечением множества различных специалистов, в том числе физиотерапевтов и специалистов по трудотерапии (occupational therapists – специалисты, помогающие людям с ограниченными возможностями освоиться в жизни) – это лучший способ контроля мышечной дистрофии Дюшенна.
Иметь доступ к специалистам из разных областей – жизненно важно для обеспечения человека с мышечной дистрофией Дюшенна всей необходимой комплексной поддержкой. Это означает, что у пациента должна быть возможность за одно посещение специализированного центра проконсультироваться у специалистов разных направлений: получить консультации по дыхательной системе, кардиосистеме и физиотерапии. Работая вместе, такие специалисты обеспечат более эффективный уход.
Важно регулярно проверяться у специалистов, чтобы принимать решения о новых методах лечения в самое подходящее время, и возможно, предвидеть и предотвращать нарушения. Рекомендуется посещать Вашего лечащего врача раз в шесть месяцев, а физиотерапевта – каждые три-четыре месяца.
Физиотерапевт даст рекомендации касательно вмешательств (таких как упражнения для развития гибкости), которые могут потребоваться. Важно позволить Вашему сыну быть как можно активнее, а Ваш физиотерапевт будет Вам помогать.
Стероиды (преднизон или дефлазакорт) часто назначаются при мышечной дистрофии Дюшенна, так как они замедляют на какое-то время процесс ослабевания мышц и нарушения подвижности и предотвращают или откладывают развитие осложнений. Однако есть множество возможных побочных эффектов, которые необходимо тщательно контролировать.
Начали появляться другие лекарства для мышечной дистрофии Дюшенна, включая Трансларну (аталурен), которая на данный момент доступна в некоторых европейских странах и способна замедлить развитие симптомов у мальчиков с мышечной дистрофией Дюшенна. Трансларна действуют только на небольшую группу мальчиков, которые являются носителями определенной мутации в гене дистрофина («нонсенс» мутация, где точечная мутация в последовательности ДНК приводит к появлению стоп-кодона). Ваш лечащий врач сообщит Вам, будет ли это лекарство эффективным для Вашего сына. В скором времени могут быть одобрены и другие средства для определённых мутаций.
Продолжается интенсивное изучение возможного лечения мышечной дистрофии Дюшенна. Некоторые лекарства сейчас проходят клинические испытания.
Полезно иметь копию генетического отчета (с типом и расположением мутации в гене дистрофина, обнаруженной у Вашего ребенка). Это поможет определить, какое лекарство или испытание больше подходит для Вашего ребенка.
Реестр мышечной дистрофии Дюшенна предоставит свежую информацию о ходе клинических испытаний и может помочь определить, какие дети предположительно могут быть допущены к конкретным клиническим исследованиям. Ваши врачи расскажут Вам как зарегистрировать Вашего ребенка в этом реестре.
The North Star Adult Network, в которую входят эксперты-консультанты по нервно-мышечным заболеваниям, специалисты смежных профессий, сами пациенты, живущие с мышечной дистрофией Дюшенна, и организация Muscular Dystrophy UK, работает над улучшением стандартных методов ухода и поддержки для взрослых на территории Соединенного Королевства. Есть и педиатрическая версия – the North Star Project – которая работает над улучшением ухода за детьми с мышечной дистрофией Дюшенна.
Если Вы хотите узнать больше о последних исследованиях команды, свяжитесь с научно-исследовательским отделом по телефону: 020 7803 4813 или по почте: [email protected]
Характеристика заболевания
Заболевание носит название своего первооткрывателя – Дюшена, который смог доказать генетическую природу. Миопатия Дюшена не
относится к распространенным патологиям, так как встречается один случай на 3,5 миллиона рожденных детей. Диагностируется исключительно у мужской половины населения в возрасте от 1,5 до 3 лет. Заболевание быстро прогрессирует.
При развитии синдрома происходит процесс гипотрофии мышц, который имеет восходящий характер. В первую очередь затрагиваются мышцы таза и проксимальный отдел ног, затем патология начинает распространяться на мышцы спины и плечи, завершающая стадия затрагивает верхние конечности.
Но стоит учитывать и тот факт, что заболевание оказывает негативное влияние и на иные отделы тела:
- деформация позвоночника;
- наблюдается искривление стоп и грудной клетки;
- заболевания сердца.
- у пациента наблюдается дебильность, которая затрагивает третью часть больных.
Что касается прогноза при миодистрофии Дюшенна, то он не очень благоприятный, так как наблюдается интенсивное прогрессирование синдрома.
Ребенок постепенно перестает ходить к десятилетнему возрасту. Летальный исход наступает в результате инфицирования дыхательных путей или остановки сердца.
Тип наследования
Синдром Дюшена относится к заболеваниям, которые передаются по наследству. К носителям патогенного гена относятся женщины.
Наследуется по рецессивному типу, сцепленному с Х-хромосомой. Большинство случаев, при которых была выявлена миопатия, были спровоцированы новыми мутационными генами. Данный вид заболевания относится к быстропрогрессирующему и злокачественному.
Клиника синдрома
Миопатия Дюшенна начинает проявляться у мальчиков в возрасте с 1,5 лет, к первым симптомам относятся:
- нарушения двигательной функции, пациенту сложно стоять, он испытывает неловкость;
- мальчик часто спотыкается, падает во время прогулок, как следствие развивается двигательная пассивность;
- ребенку составляет трудность подняться вверх по лестнице, походка похоже на «утиную»;
- ребенку тяжело подниматься с кровати или со стула;
- наблюдается гипертрофия икроножных мышц, данный признак относится к наиболее яркому, который сразу свидетельствует о прогрессировании заболевания;
- заболевания сердца;
- нарушения в биоптатах мышц скелета;
- контрактура крупных суставов, деформация стопы наблюдается у пациента в результате интенсивного развития патологии;
- в возрасте десяти лет ребенок перестает передвигаться самостоятельно и становиться прикованным к инвалидному креслу;
- в возрасте 14 лет ребенок становиться полным инвалидом.
Если были выявлена выше представленная симптоматика стоит немедленно пройти обследование и лечение. В данной ситуации не стоит тратить ни минуты, так как от этого зависит жизнь пациента.
Постановка диагноза
В первую очередь пациента осматривают, чтобы выявить внешние проявления болезни Дюшенна. Затем ребенка направляют на дополнительное обследование, которое заключается в следующем:
- электрокардиограмма помогает выявить поражение миокарды латеральное и заднее-нижней стенок левого желудочка;
- определение уровня дистрофина в ткани мышц;
- биохимическое исследование крови, помогает выявить активность фермента креатинфорсфокиназы;
- в обязательном порядке проводиться генетическая диагностика;
- проводиться электронейромиография, помогает выявить некроз мышечной ткани;
- биопсия мышечной ткани, считается главным методом диагностирования миопатии.
На основании полученных результатов ставиться окончательный диагноз, и назначается терапия.
Лечение: цели, методы, трудности
Лечение миопатии Дюшенна невозможно, можно только продлить время жизни и поддержать состояние пациента на стабильном уровне.
Различные методы современной медицины помогают облегчить состояние, замедлить прогрессирование болезни. Ниже будут представлено лечение миодистрофии Дюшена для разных возрастов, но бывают такие случаи, когда требуется применять сразу несколько способов терапии:
- Пациент в возрасте до 5 лет. В данной ситуации не требуется радикальное лечение. Рекомендуется осведомить родителей о заболевании, рассказать о проблемах и последствиях, рассказать, какие нагрузки допустимы для ребенка, и провести в обязательном порядке генетическую консультацию.
- Пациент в возрасте до 8 лет. Большинству детей в таком возрасте уже требуется поддержка мускулатуры нижних конечностей. Кортикостероиды помогают замедлить ход развития заболевания на некоторое время. Рекомендуется принимать Преднизолон или Дефлазакорт.
- Пациент в возрасте от 8 лет до 20 лет. Наблюдается постепенное ослабление мышц, которое в результате заставляет ребенка прибегнуть к инвалидному креслу.
- Пациент в возрасте от 20 лет. Препараты в данной ситуации помогут только облегчить состояние, начинает прогрессировать инфекция дыхательных путей.
В течение всей жизни пациент должен принимать препараты, которые обеспечивают поддержку организма:
- витамины В и Е, аминокислоты, кальций, анаболические гормоны, калия оротат;
- Прозерином, Галантаминон, Оксазилон.
Также курсами проводят инъекции Ретаболила, АТФ, Цереброзилил, Анарилин. Рекомендуется принимать глютаминовую кислоту, Ексазил.
Параллельно проводиться лечение ЛФК, которое осуществляется непродолжительными курсами с перерывами. Также рекомендуется делать массаж, электрофорез Прозерина, Липазы, кальция хлорида, применяют лечебные ванны и индуктотермию. В особо тяжелых случаях лечение проводиться в домашних условиях. Чтобы продлить жизнь на несколько лет можно попробовать глюкокортикоидное лечение.
Параллельно пациент должен постоянно находиться под наблюдением кардиолога. Также обязательно ребенок должен полноценно питаться. В рацион включены: растительные жиры и белковая пища, свежие или приготовленные на пару овощи, фрукты, молочные продукты, овсянка, яйца, мед, орехи и морковь. Не стоит потреблять крепкий чай или кофе, спиртные напитки, приправы, сахар, капусту и картофель.
Осложнения и последствия
Как уже говорилось ранее, данное заболевание сильно укорачивает жизнь человека и это можно считать самым серьезным последствием. Так как патология быстро прогрессирует, то мышцы становятся слабыми, возникают необратимые последствия в работе сердца, наблюдается сбой дыхательной системы.
Если миопатия Дюшена будет своевременно выявлена и лечение будет подобрано правильно, то пациент сможет дожить до 30 лет.
К осложнениям можно отнести такие заболевания и патологии:
- Остеопороз. Чтобы постараться отсрочить данную патологии рекомендуется принимать витамин Д и кальций, поэтому стоит скорректировать питание. При остеопорозе принимают биосфосфонаты.
- Заболевания суставов и позвоночника. Суставы становятся практически полностью подвижными, поэтому рекомендуется носить шины. В некоторых случаях проводят операции.
- Заболевания пищеварительной системы и проблемы с весом. С возрастом человек начинает терять в весе из-за некроза
 мышц, поэтому требуется постоянная консультация диетолога. Также пациента может потревожить и запор, который также является следствием малоподвижного образа жизни. В данной ситуации рекомендуется принимать слабительные препараты, больше есть продуктов, которые богаты на клетчатку. В возрасте от 17 лет у больного могут возникнуть проблемы с жевательной и глотательной функцией. В редких случаях проводиться гатростомия.
мышц, поэтому требуется постоянная консультация диетолога. Также пациента может потревожить и запор, который также является следствием малоподвижного образа жизни. В данной ситуации рекомендуется принимать слабительные препараты, больше есть продуктов, которые богаты на клетчатку. В возрасте от 17 лет у больного могут возникнуть проблемы с жевательной и глотательной функцией. В редких случаях проводиться гатростомия. - Нарушения дыхательной функции. Из-за слабого кашлевого механизма могут прогрессировать респираторные инфекции, так как бактерии и слизь не выводятся. В качестве профилактики рекомендуется делать прививки. Также по мере прогрессирования патологии у пациента наблюдается снижение уровня кислорода в организме, как следствие возникают утренние головные боли, бессонница, слабость, нервная возбудимость, особенно во сне.
- Заболевания сердца. В большинстве случаев развивается кардиомиопатия.
Профилактика болезни затрудненная, так как данная патология является генетической, поэтому нужно уделить внимание консультации у генетиков тем семьям, у которых наблюдается отягощенная наследственность.
Мышечная дистрофия Эмери-Дрейфуса. Причины
Х-хромосомная рецессивная мышечная дистрофия Эмери-Дрейфуса вызывается мутациями в гене EMD (расположен на Х хромосоме), кодирующем белок эмерин. На сегодня известно о более 70 уникальных мутациях, из них наиболее часто встречаемыми являются точечные мутации, небольшие делеции, инсерции или те, которые обычно приводят к стоп-кодонам. У людей, с такими мутациями, белок эмерин чаще всего отсутствует полностью, но, в некоторых случаях, белок может присутствовать, но в дефиците. Эмерин является белком, который принадлежит к семейству ядерных белков, эти белки считаются важными в поддержании структуры ядерной мембраны. Белок эмерин не является необходимым для модели выживания клеток, так как некоторые животные, которые имеют дефицит белка, не имеют явной мышечной миопатии. Также стоит обратить внимание на то, что в некоторых случаях, аутосомно-доминантная и аутосомно-рецессивная мышечная дистрофия Эмери-Дрейфуса также может быть вызвана мутацией в хромосоме 1, а точнее в гене, который кодирует белок ламин A/C (LMNA). Интересно отметить то, что одинаковые мутации могут привести к различным фенотипам мышечной дистрофии Эмери-Дрейфуса между братьями и сестрами. У одного человека могут быть только мягкие проявления, в то время как у других лиц могут проявлять очень тяжелые признаки болезни. Этот аспект указывает на то, четкая корреляция между клиническим фенотипом и типами мутаций отсутствует.
Мышечная дистрофия Эмери-Дрейфуса. Патофизиология
Белок эмерин присутствует в большом комплесе факторов, которые учавствуют в построении и в поддержании нуклеоскелета и цитоскелета. Этот комплекс белков включает в себя белки ядерной мембраны, к ним относятся эмерин, ламин A/C, SUN1, SUN2, несприн-1, несприн-2 и большое количество других, вспомогательных белков, которые создают механическую связь между нуклеоскелетом и цитоскелетом. Мутации в гене EMD могут происходить по всей длине гена и это почти всегда приводит к полному отсутствию этого белка в клетках мышц. В редких случаях, у некоторых пациентов этот белок может производиться, но в значительно уменьшенных количествах. Белок эмерин присутствует почти во всех типах клеток нашего организма, хотя его высшее выражение проявляется именно в скелетных мышцах и в мышцах сердца. Белок способен связываться со многими ядерными белками, в том числе с некоторыми ген-регуляторными белками, неспринами (белки, которые действуют в качестве молекулярного каркаса), F-актином и с ламинами. Поэтому, отсутствие этого белка всегда приводит к разрушению структуры клеток мышц, что в свою очередь, почти всегда приводит к развитию проявлений этой болезни.
Мышечная дистрофия Эмери-Дрейфуса. Симптомы и проявления
Следующая триада симптомов и проявлений может навести на мысль о наличии у человека мышечной дистрофии Эмери-Дрейфуса:
- Медленно прогрессирующая мышечная слабость
- Ранние контрактуры в локтевой, голеностопной области и в области шеи
- Аномалии в проводящей системе сердца.
Мышечная дистрофия Эмери-Дрейфуса начинает развиваться и проявляться, как правило, уже в подростковом возрасте, но она также может начать проявляться даже у лиц в возрасте за тридцать лет. Пациенты, как правило, развивают слабость малоберцовых мышц и это приводит к проблемам в ходьбе уже в конце первого десятилетия или в начале подросткового возраста.
Контрактуры
Контрактуры (ограничения движений в суставах) часто развиваются еще до слабости. Эти контактуры могут развиться в:
- Локтевых суставах
- В позвоночнике
- В лодыжке
Слабость
- Симметричная слабость бицепсов, трицепсов и малоберцовых мышц
- Слабость в лицевых, бедренных мышцах, а также в руках
Проблемы с сердцем (почти универсальные проявления)
- Сердечная болезнь, как правило, начинается после наступления слабости в мышцах и она может проявляться в виде обмороков уже в возрасте 20 лет.
- Кардиостимуляторы часто необходимы при достижении пациентом возраста 30 лет.
- Сердечная болезнь может также стать причиной внезапной сердечной смерти.
- Брадикардия, аритмия предсердий, дефекты проводимости и мерцательный паралич также фиксировались у пациентов с этой дистрофией.
- Поздние проблемы могут включать кардиомиопатии предсердий или желудочков.
- Что касается женщин, то 10-20% от всех женщин с дистрофией, имеют аритмии предсердий или нарушения проводимости, которые должны регулярно мониториться на ЭКГ. Постоянные проверки могут предотвратить развитие внезапной сердечной смерти.
Мышечная дистрофия Эмери-Дрейфуса. Диагностика
Лабораторные анализы
Уровни креатининкиназ повышенны не более чем в 10 раз от нормальных уровней, в большинстве случаев. Однако, если уровень белка будет крайне повышен, то медицинский персонал должен провести обследование человека на другие нарушения, в том числе это касается дистрофии Дюшенна/Беккера.
Электромиография (ЭМГ) и исследование нервной проводимости (ИНП)
- ЭМГ и ИНП должны быть выполнены для подтверждения миопатической природы заболевания и для исключения других нервно-мышечных синдромов.
Электрокардиограмма (ЭКГ)
- ЭКГ должна выполняться у всех пациентов с мышечной дистрофией Эмери-Дрейфуса.
- Ранние изменения включают низкие амплитуды волны P и длительный интервал PR.
- Классический образец сердечных проявлений болезни включает в себя ритм 40-50 ударов в минуту без Р-волн.
Процедуры
Биопсия мышц должна быть проведена у всех пациентов с предполагаемой дистрофией для рутинного гистологического окрашивания. При проведении иммуногистохимических исследований, антитела к белку эмерин могут помочь в подтверждении диагноза.
Гистология
Гистохимические пятна могут показывать типичные миопатические аспекты, в том числе изменчивость в размере мышечных волокон с наличием маленьких круглых волокон и иногда с некротическими и регенерирующимися волокнами. Небольшое увеличение эндомизиальной соединительной ткани и внутренних ядер часто присутствуют у пациентов с мышечной дистрофией.
Иммуногистохимическое окрашивание с использованием антител антиэмерин может показать отсутствие нормальной окраски внутренней ядерной мембраны. Аналогичная картина получается и при окрашивании периферических лейкоцитов, фибробластов кожи и щечных клеток.
Мышечная дистрофия Эмери-Дрейфуса. Лечение
Специфической лечебной терапии мышечной дистрофии Эмери-Дрейфуса не существует, но, медицинскому персоналу очень важно постоянно проводить интенсивные терапии, это поможет в сохранении мышечной активности, это обеспечит максимальную функциональную способность и увеличит продолжительность жизни.
Основной задачей таких подходов является предотвращение внезапной сердечной смерти.
- Кардиостимуляторы должны быть имплантированны в больных с брадикардией.
- Церебральная эмболизация, тромбообразование и кардиомиопатия могут по-прежнему возникать даже у пациентов с кардиостимулятором.
- Трансплантацию сердца следует рассматривать у пациентов с прогрессирующей неизлечимой кардиомиопатией.
- Желудочковые аритмии могут возникнуть в конце болезни и по этой причине кардиовертер-дефибрилятор может быть предпочтительнее простого кардиостимулятора.
Другой основной проблемой является профилактика и коррекция скелетных аномалий (контрактуры) и поддержание способности к передвижению.
- Тенотомия ахиллова сухожилия может помочь в стабилизации контрактур лодыжки.
- Вопросы с контактурами шеи и позвоночника можно решить хирургическим вмешательством.
Агрессивное использование пассивного растяжения, фиксации и ортопедические процедуры могут позволить пациенту оставаться активным как можно дольше. Как и при других наследственных миопатиях, командный подход, в том числе участие невропатолога, пульмонолога, кардиолога, хирурга-ортопеда, физиотерапевта, физиотерапевта, ортопеда и консультантов, обеспечивает наилучшие результаты лечения.
Хирургический подход
- Цель хирургического подхода должна состоять в том, чтобы пациент был мобильным по максимуму и до тех пор, насколько это возможно.
- Ортопедические операции могут быть необходимы в исправлении или в предотвращении развития контрактур и в повышении диапазона движений.
Мышечная дистрофия Эмери-Дрейфуса. Осложнения
- Предсердные сердечные нарушения проводимости, которые могут вылиться в обморок или в внезапную смерть.
- Тяжелые контрактуры могут вызвать значительные ортопедические проблемы.
Мышечная дистрофия Эмери-Дрейфуса. Прогноз
- Внезапная сердечная смерть является частой причиной ранней смерти.
- Мышечная дистрофия Эмери-Дрейфуса является прогрессивным расстройством и пациенты часто умирают в середине взрослой жизни от прогрессирующей легочной или сердечной недостаточности.
Наследственные миопатии это неоднородная группа заболеваний.
Врачу-невропатологу важно установить гетерозиготное носительство гена миопатии, прежде чем дать совет в отношении деторождения у ближайших родственников больных миопатией.
В некоторой степени здесь помогает выявление некоторых неврологических симптомов, а также электрофизиологическое и биохимическое исследование.
Неврологическими микросимптомами гетерозиготности являются укорочение брюшка икроножной мышцы и удлинение ахиллова сухожилия, снижение или отсутствие ахилловых и коленных рефлексов.
При электромиографическом исследовании накожными электродами у больных и гетерозиготных носителей отмечается снижение вольтажа кривой ЭМГ, а в сыворотке крови — повышенное содержание альдолазы и креатинкиназы. Скрининг па миопатию Дюшенна проводится у детей 5—6-дневного возраста путем определения концентрации креатинкиназы в капле крови. Концентрация креатинкиназы у данных детей повышена.
Непрогрессирующие миопатии
С внедрением в практику электронномикроскопического исследования выявлены непрогрессирующие врожденные формы миопатий. Для них характерны раннее начало заболевания, большая слабость мышц проксимальных отделов конечностей и стационарное (непрогрессирующее) течение. Выделяют несколько форм врожденных видов болезни.
Миопатия центрального стержня. В центре мышечного волокна отсутствует активность мышечной фосфорилазы, молочной дегидрогеназы, что выявляется гистохимически, а при электронно-микроскопическом исследовании — дезорганизация фибрилл — нечеткость деления на саркомеры и изменение полос. Мышечное волокно напоминает картину волокна, наблюдаемую при денервации.
Нитевидная миопатия.
При гистологическом исследовании во многих мышечных волокнах есть нитевидные структуры, расположенные в центре или на периферии мышечных волокон, в которых отсутствует поперечная исчерченность волокон и разный их калибр.
Клинически у больных кроме снижения мышечного тонуса, истончения мышц выявляются аномалии скелета, проявляющиеся деформацией позвоночника и грудной клетки, и вытянутость лицевого черепа.
Миотубулярная (центральноядерная) миопатия. У больных, как и при предыдущей форме, отмечаются понижение мышечного тонуса конечностей и туловища, опущение век, наружная офтальмоплегия, а также выявляются небольшие мышечные волокна с расположенными в центре ядрами, которые окружены светлым полем цитоплазмы фибрилл.
Миопатия с гигантскими митохондриями. Проявляет себя врожденной гипотонией мышц, а при гистологическом исследовании выявляются гигантские митохондрии под сарколеммой, между миофибриллами.
Миопатические синдромы наблюдаются также при некоторых наследственных и ненаследственных заболеваниях: тиреотоксикозе, гиперкортицизме, акантоцитозе, полимиозите, гликогенозе Мак-Ардла, бронхогенном раке.
Источник: http://surgeryzone.net/bolezni/vrozhdennye-miopatii.html
Что такое дистрофия или миопатия Дюшенна
Это наиболее распространенный тип дистрофии мышц, поражающий примерно 1 из 4 000 мужчин. Из-за врожденного дефекта гена дистрофина мышечная ткань утрачивает свою структуру и не развивается. Мышцы не могут двигаться и сокращаться, что приводит к необратимым дегенеративным изменениям в опорно-двигательном аппарате и деформации скелета. Недуг быстро прогрессирует, сопровождается нарушениями в работе сердца, органов дыхания, эндокринной и нервной систем.
Больной с детского возраста испытывает трудности с передвижением. Он имеет специфическую походку и осанку, позже сверстников начинает ходить и делает это хуже. Уже с 8-10 лет пациенту требуются костыли, с 12 — инвалидная коляска. Больные старше 16 лет не только не могут передвигаться самостоятельно, но и испытывают проблемы с дыханием, интеллектом, сердцем. Смерть в среднем наступает в 30 лет, однако есть люди, живущие дольше.
Болезнь имеет код по мкб 10 — G71.0, часто определяется как миопатия Дюшенна-Беккера, однако это неверно. Существует отдельная патология — прогрессирующая мышечная дистрофия Беккера. Она отличается медленным течением и поздними проявлениями. Симптомы возникают исключительно в возрасте 15-20 лет, в то время как при болезни Дюшенна юноши в данный период уже прикованы к инвалидной коляске. Продолжительность жизни при миопатии Беккера тоже больше. Таким образом, болезнь Дюшенна-Беккера — это не единая патология, а два разных состояния.
Также диагностируют амиотрофию Дюшенна, приводящую к нарушению в работе мышц. Однако это не самостоятельная патология, а группа прогрессивных спинальных заболеваний, поражающих взрослых.
Причины дистрофии Дюшенна
Причиной появления заболевания является врожденный дефект половой X-хромосомы. Поражен тот ген, который отвечает за выработку в организме мышечного белка — дистрофина. Именно он является основой всех мышечных волокон на клеточном уровне. Вещество необходимо для корректного развития скелета, способности мышц сокращаться и расслабляться множество раз подряд.
При болезни Дюшенна белок отсутствует вовсе или является дефектным, то есть не выполняет свою функцию. Он заменяется жировой или соединительной тканью, из-за чего движение становится невозможным.
Заболевание имеет рецессивный тип наследования, сцепленный с X-хромосомой, и возникает при мутациях в обеих парных носителях. Патология поражает преимущественно мальчиков, поскольку в их генетическом наборе имеется одна X-хромосома. Если она дефектная, то болезнь настает.
Патогенез во время мышечной дистрофии Дюшенна
Миодистрофия Дюшенна обусловлена мутацией в гене дистрофин, отвечающего за выработку одноименного белка. Из-за этого не происходит соединения цитоскелета каждой клетки мышечных волокон с основной базальной пластиной. Кроме того, избыток кальция проникает в клеточную мембрану. Митохондрии наполняются водой и разрываются, их разрушение приводит к отмиранию целой мышечной клетки. Те волокна, которые подверглись некрозу, больше не выполняют свою функцию и состоят теперь из иных тканей.
Симптомы прогрессирующей мышечной дистрофии Дюшенна
Главным проявлением болезни является мышечная слабость, перерастающая в атрофию. Это касается скелетных мышц, расположенных в бедрах, тазу, плечах, ногах. Позже поражаются шея, руки и прочие части тела. Кроме того, симптомами болезни называют:
- неуклюжую походку;
- повышенный тонус икроножных мышц;
- хроническую усталость;
- невозможность бегать или прыгать;
- поясничный лордоз;
- расстройства неврологического и психического характера;
- трудности с концентрацией;
- ухудшение памяти;
- сколиоз;
- гормональные нарушения;
- потерю способности самостоятельно ходить.
Заболевание имеет две формы: начальную и прогрессирующую. Они неизбежно связаны друг с другом: одна перетекает в другую со временем. Известно, что миопатия Дюшенна, сопровождающаяся дистрофией мышц на начальной стадии, заметна у мальчиков уже в возрасте 2 лет. Прогрессировать заболевание начинает к 8-12 годам.
Мышечная дистрофия Дюшенна
Прежде всего у пациента отмечают сильное увеличение икроножных мышц в размере. Такое явление нечасто называют псевдогипертрофическая мышечная дистрофия Дюшенна. Это провоцирует затруднения при ходьбе и частые падения: больной вынужден передвигаться, опираясь на кончики пальцев, а не на всю стопу. Кроме того, он испытывает:
- боли во всех скелетных мышцах;
- трудности во время монотонного сидения или стояния;
- скованность движений.
Важно! С самого начала болезнь отрицательно влияет на способность человека нормально запоминать и воспроизводить даже простейшую информацию. Из-за этого ребенок опаздывает в развитии, плохо разговаривает, не может сконцентрироваться.
Прогрессирующая дистрофия Дюшенна
На поздних стадиях болезни человеку трудно глотать, ему требуется специальная трубка для приема пищи. Причем самостоятельные попытки проглотить еду приводят к угрозе возникновения аспирационной пневмонии. Кроме того, у пациента наблюдаются:
- неспособность передвигаться;
- уменьшение размера мышц и сухожилий;
- затруднение дыхания, нередко пациент дышит только с медицинской помощью;
- сильное искривление позвоночника;
- поражение мышц сердца и, как следствие, серьезные перебои в его работе.
Методы диагностики заболевания Дюшенна-Беккера
Диагностикой болезни занимается невролог. Для подтверждения наличия патологии проводят ряд исследований:
- Тест ДНК. Позволяет с точностью определить наличие мутации гена.
- Биопсия мышечных тканей. Используется редко, заключается в заборе образца мышечных волокон и определение наличия дистрофина при помощи специального красителя.
- Пренатальное тестирование. Проводится на 11 неделе беременности с целью выявить риск нервно-мышечных нарушений, особенно если один или оба родителя являются носителями.
- Забор крови плода. Осуществляется на 18 неделе беременности, применяется в крайних случаях врачом-генетиком.
Внимание! Будущим родителям следует быть осторожными с тестами во время беременности. Однако если такая необходимость возникла, то предпочтение стоит отдавать раннему тестированию, чтобы минимизировать вероятность выкидыша.
Лечение мышечной дистрофии Дюшенна
Эффективных препаратов для избавления от миодистрофии Дюшенна не существует. Лечение, как правило, симптоматическое, позволяющее облегчить состояние больного. Оно включает в себя:
- прием кортикостероидов: преднизолона и дефлазакорта;
- употребление бета 2-агонистов, замедляющих прогресс болезни;
- умеренную физическую активность, например, плавание;
- физиотерапию;
- использование инвалидной коляски и ортопедических приспособлений во время сна;
- ношение респираторов, облегчающих дыхание.
В современной медицине практикуется лечение стволовыми клетками, которыми заменят поврежденные миоциты. Инъекции со здоровыми клетками делают во все мышцы через каждые 2 мм: эта процедура отнимает очень много времени.
Известны также стволовые клетки под названием перициты, располагающиеся в кровеносных сосудах скелетных мышц. Ученые получают их небольшое количество, выращивают искусственным путем и вводят в кровоток. Некоторые из них попадают в мышцы и заполняют их. Эффективность операции однозначно не определена учеными. 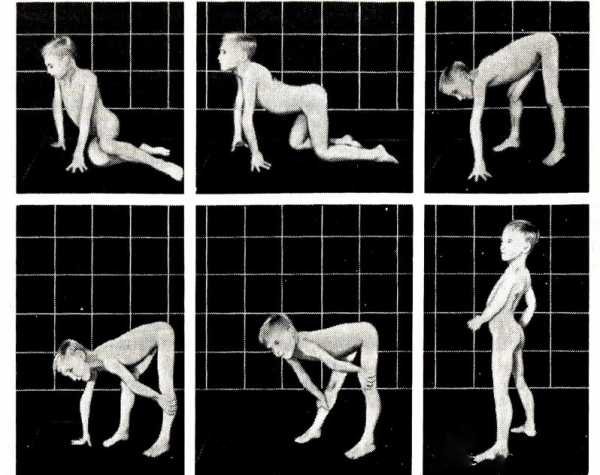
Для профилактики осложнений болезни пациент должен вести здоровый образ жизни, правильно питаться, достаточное время находиться на свежем воздухе. Запрещается ограничивать передвижения человека и держать его на постельном режиме. Без хотя бы минимальных попыток ходьбы мышцы разрушаются очень быстро.
Из-за того, что при дистрофии Дюшенна быстро повреждаются не только скелетные мышцы, но и сердце, органы дыхания, человек не может долго жить в таком состоянии. Даже при условии приема препаратов, облегчающих состояние, больные редко доживают до 35 лет. Больше 50% из них умирают еще в подростковом возрасте, без соответствующей помощи — еще быстрее.
При искусственной вентиляции легких, приеме поддерживающих лекарственных средств для сердца, использовании инвалидной коляски и трубки для кормления некоторые пациенты проживают 40-50 лет. Однако они нуждаются в постоянном уходе и наблюдении, так как не могут самостоятельно выполнить элементарные гигиенические процедуры и даже попросить о помощи.
Вылечить миодистрофию Дюшенна невозможно, однако облегчить состояние больного помогут лекарственные и ортопедические средства. Из-за значительных поражений внутренних органов пациенты обычно погибают в молодом возрасте. Но современные достижения в области применения стволовых клеток дают возможность надеяться на увеличение средней продолжительности жизни у людей, страдающих от этой болезни.
healthage.ru
Миопатия Дюшена: Диагноз. Лечение. Осложнения.
Мышечная дистрофия Дюшенна (МДД) или миопатия Дюшена — это врожденное заболевание, которое вызывает прогрессирующую слабость мышц. Это заболевание проявляется в детстве — родители ребенка могут заметить, что ему трудно стоять, бегать или карабкаться на лесенки на детских площадках. Болеют этим видом миопатии преимущественно мальчики, но девочки могут быть носителями так называемого гена Дюшена (известны крайне редкие случаи, когда от этого заболевания страдали и девочки). Пациентам с таким диагнозом приходится часто обследоваться у врача, а с определенного возраста (в среднем — с девяти лет) у них возрастает потребность в средствах для облегчения признаков этого нарушения.
Что такое миопатия Дюшена
Это серьезное заболевание поражает, главным образом, мышцы в области туловища, бедер и плеч. При этом пациенты, как правило, могут свободно использовать руки и пальцы, но у них возникают проблемы с ходьбой, бегом, и так далее. Мышечная слабость прогрессирует постепенно. Обычно она проявляется в раннем детстве, но поначалу симптомы миопатии Дюшена выражены очень слабо. С возрастом они становятся все более выраженными, и приводят к резкому снижению качества жизни.
Миопатия Дюшена диагностируется приблизительно у одного из 3500 мальчиков.
В мышечной ткани содержится дистрофин — белок, необходимый для нормальной работы мышц. У людей с миопатией Дюшена этого вещества слишком мало. Со временем это приводит к повреждению мышечных волокон и ослаблению мышц. Причиной этого является особый ген, который передается от родителей к детям, либо генные мутации, произошедшие в период внутриутробного развития.
Для каждого сына женщины, которая является носителем гена Дюшена, вероятность развития миопатии Дюшена составляет ровно 50%. Дочери такой женщины станут носителями этого гена с такой же вероятностью.
Если у ребенка миопатия Дюшена, значит ли это, что у кого-то из членов семьи есть ген Дюшена? Не обязательно. Приблизительно в половине случаев заболевшие миопатией этого типа не получают дефектный ген от одного из родителей. В клетках плода еще во время беременности происходят мутации, результатом которых и становится миопатия. Это может произойти из-за «ошибки», которая случилась при копировании родительских генов в клетки, которые должны будут образовать организм ребенка. Почему это происходит, в настоящее время неизвестно.
Точно узнать, есть ли у кого-то из членов вашей семьи ген Дюшена, можно, только при помощи генетического консультирования.
Каковы симптомы?
Обычно первые симптомы миопатии Дюшена появляются в возрасте 1-3 лет. Родители могут заметить следующие признаки миопатии Дюшена:
- Ребенку трудно ходить, бегать, прыгать, подниматься по лестницам. Походка ребенка может отличаться от походки его ровесников — он ходит вразвалочку, и менее уверенно, чем остальные. Иногда дети с миопатией Дюшена начинают ходить позже остальных, однако и совершенно здоровые малыши иногда делают первые шаги несколько позже сверстников;
- В более старшем возрасте ребенок может опираться на руки, чтобы встать;
- У ребенка могут наблюдаться проблемы с обучением — как правило, не очень серьезные.
Иногда первым признаком миопатии Дюшена является замедленное развитие речи.
Как диагностируется
В первую очередь врачи, как правило, просто наблюдают за ребенком, в особенности за тем, как он ходит, бегает и встает с пола. Если основания подозревать миопатию Дюшена, будет назначен анализ крови на креатинкиназу — это фермент, уровень которого у людей с этим нарушением всегда очень высок (в 10-100 раз выше нормы). Если уровень креатинкеназы у ребенка в норме, миопатию Дюшена исключают и начинают искать другие причины появившихся у малыша симптомов.
Следующим этапом диагностики миопатии Дюшена является биопсия мышечной ткани и/или генетическое тестирование.
В ходе биопсии врач берет небольшой фрагмент мышечной ткани для дальнейших анализов; процедуру проводят под общей анестезией. Образец ткани изучают под микроскопом при помощи особых техник, чтобы оценить состояние мышечных волокон и количество дистрофина.
Для проведения генетического тестирования необходимо необходимое количество крови пациента. С помощью этого метода выявляют гены, которые отвечают за развитие миопатии Дюшена. В большинстве случаев этот способ позволяет точно диагностировать данное заболевание.
Лечение миопатии Дюшена
В настоящее время вылечить эту болезнь невозможно. Однако существует ряд способов, которые могут облегчить состояние пациента на разных стадиях развития миопатии и, возможно, несколько замедлить прогрессирование заболевание.
Ниже описаны способы лечения для определенных возрастных групп, но нередко при лечении одного пациента приходится сочетать сразу несколько методов терапии.
Пациенты дошкольного возраста
Обычно в этом возрасте детям с миопатией Дюшена еще не нужно лечение. Родителям могут предложить:
- Подробную информацию о миопатии Дюшена. Врач проведет с родителями беседу и подробно расскажет о том, как эта болезнь будет влиять на состояние ребенка, и какова ожидаемая продолжительность жизни пациента (большинство людей с таким диагнозом доживают лишь до 20-25 лет). При желании родители могут обратиться к другим специалистам (например, к психологу), а также в группы поддержки;
- Рекомендации относительно допустимых физических нагрузок для ребенка;
- Генетическую консультацию для членов семьи. Многие родители желают узнать, являются ли они носителями гена Дюшена. Это особенно важно для тех, кто в будущем планирует снова родить ребенка.
Уже в дошкольном возрасте ребенка начинают регулярно обследовать, чтобы, когда это будет необходимо, можно было вовремя начать лечение.
Пациенты в возрасте 5-8 лет
Детям такого возраста может потребоваться поддержка для мышц ног. Например, им могут рекомендовать надевать на ночь шины для лодыжки или более длинные шины, для голени.
При помощи кортикостероидов можно замедлить развитие миопатии и в течение некоторого времени сохранять мышцы достаточно сильными. Пациенты постоянно или курсами принимают такие препараты, как преднизолон или дефлазакорт. Поскольку кортикостероиды могут вызывать серьезные побочные эффекты, ребенок обязательно должен наблюдаться у врача.
Пациенты от 8 лет до позднего переходного возраста
Через какое-то время после достижения ребенком возраста восьми лет его мышцы начинают заметно слабеть. Ходить со временем становится все труднее пока, наконец, ребенку не придется начать передвигаться на инвалидном кресле. Возраст, в каком это происходит, варьируется от пациента к пациенту. Часто это случается между 9 и 11 годами, но дети, которые достаточно рано начали принимать кортикостероиды, иногда могут продолжать ходить несколько дольше.
Вскоре после того, как ребенку для передвижения становится необходимым инвалидное кресло, у него начинают развиваться и другие осложнения, поэтому ему могут потребоваться более частые обследования. Все осложнения необходимо начинать лечить как можно раньше.
Кроме этого, родителям нужно позаботиться о практической стороне жизни ребенка — на первых порах помогать ему передвигаться в кресле, а также по возможности приспособить свой дом под его нужды.
Пациенты от позднего переходного возраста до 20+ лет
В этом возрасте мышечная слабость вызывает все больше проблем, и пациенту все чаще требуется помощь других людей. Увеличивается вероятность развития тяжелых осложнений, таких как легочные инфекции.
Прогноз
Как говорилось выше, миопатия Дюшена — тяжелое заболевание, которое значительно сокращает жизнь человека. Со временем мышечная слабость вызывает все более серьезные проблемы с дыхательной системой и работой сердца. В прошлом большинство пациентов с миопатией Дюшена доживали лишь до 20-23 лет. Сегодня все больше людей с этим диагнозом доживают до 27 лет, а иногда и до более старшего возраста. Отметим, что продолжительность жизни зависит от многих факторов, таких как сопутствующие заболевания, доступность качественной медицинской помощи, и так далее. Со временем ожидаемая продолжительность жизни при миопатии Дюшена может еще больше увеличиться.
Наиболее распространенной причиной смерти больных являются осложнения, связанные с респираторной системой, например, тяжелые инфекции дыхательных путей.
Осложнения при миопатии Дюшена и их лечение
Остеопороз. У больных миопатией Дюшена может развиваться остеопороз — заболевание, которое приводит к уменьшению плотности костной ткани. Основными причинами этого являются вынужденная малоподвижность и прием кортикостероидов. Очень важно как можно дольше предотвратить развитие этой болезни. Для этого необходимо получать достаточное количество витамина D и кальция — они требуются для того, чтоб кости оставались крепкими. Эти вещества можно получить как из пищи, так и из витаминных добавок.
Пациентам, у которых уже развился остеопороз, назначают лекарственные препараты, например, бисфосфонаты.
Осложнения, затрагивающие суставы и позвоночник
Мышечная слабость может стать причиной того, что суставы пациента со временем станут менее подвижными. В таких случаях пациентам могут рекомендовать носить шины, а иногда может потребоваться хирургическое вмешательство.
Сколиоз, или искривление позвоночника, также может стать результатом прогрессирующей мышечной слабости. Обычно он развивается уже после того, как пациент начинает передвигаться в инвалидном кресле. Чаще всего для лечения сколиоза пациенты носят специальные корсеты. Хирургические операции при этом нарушении назначают лишь в редких случаях.
Осложнения, связанные с питанием и пищеварением
У детей с миопатией Дюшена часто бывает избыточный вес, особенно если они принимают кортикостероиды. У подростков и взрослых людей с этим диагнозом, напротив, может быть дефицит массы тела из-за постепенного разрушения мышечных волокон. Чтобы удержать вес в пределах нормы, пациентам следует регулярно консультироваться с диетологами, и выполнять их рекомендации.
Запор часто становится результатом малоподвижного образа жизни. Для лечения запоров назначается прием слабительных препаратов, а для их профилактики рекомендуется употреблять в пищу больше продуктов, богатых диетической клетчаткой.
На поздних стадиях развития миопатии Дюшена — в возрасте старше 18-20 лет — у многих пациентов появляются проблемы с жеванием и глотанием пищи. В наиболее тяжелых случаях может быть необходима гастростомия — операция, в ходе которой в полость желудка вводят специальную трубку, через которую будет осуществляться кормление.
Осложнения, связанные с дыхательной системой
В подростковом возрасте дыхательные мышцы пациентов начинают ослабевать, из-за чего дыхание становится поверхностным, а кашлевой механизм становится менее эффективным. Это может привести к различным респираторным инфекциям, поскольку слизь и бактерии выводятся из дыхательных путей не так легко, как у здоровых людей. Важно лечить такие инфекции своевременно — обращаться к врачу при появлении первых симптомов, и принимать все назначенные лекарства. Для профилактики некоторых инфекционных заболеваний пациенту могут сделать прививки.
По мере того, как дыхательные мышцы слабеют, концентрация кислорода в крови снижается, особенно во время сна. Поскольку это происходит постепенно, признаки этого поначалу могут быть незаметны. Наиболее распространенными симптомами понижения уровня кислорода в крови являются головные боли по утрам, частые пробуждения по ночам, слабость, раздражительность, очень живые, насыщенные сновидения.
Своевременное обращение к врачу позволяет облегчить многие из этих симптомов, и заметно улучшить качество жизни.
Осложнения, связанные с работой сердца
У подростков и взрослых с миопатией Дюшена может развиться кардиомиопатия — нарушение, для которого характерна слабость сердечной мышцы. Признаками кардиомиопатии могут быть повышенная утомляемость, одышка, отеки ног, нерегулярное сердцебиение. Для лечения кардиомиопатии назначается медикаментозная терапия. Чем раньше ее начать, тем более эффективной она будет.
Источник http://www.womenclub.ru/
mioby.ru
Причины и симптомы миопатии Дюшена
Миопатия Дюшена — наследственная патология мышечной системы. Передается через X хромосому, проявляется нарастающей мышечной слабостью, приводящей к полной обездвиженности больного. Болеют исключительно дети мужского пола, хотя и описаны случаи заболевания девочек, но в этих случаях болезнь развивалась на фоне еще более тяжелой генетической патологии. Частота встречаемости миопатии Дюшена составляет 1 случай заболевания на 4000 новорожденных мужского пола.
Причины миопатии Дюшена
Единственна причина этого заболевания — мутация одного гена, расположенного в X-хромосоме. Данный ген отвечает за синтез белка дистрофина, обеспечивающего целостность мышечных волокон во время сокращения. При миопатии Дюшена мутация приводит к тому, что белок дистрофин вообще не синтезируется. Это становится причиной разрушения мышечных волокон, которые замещаются затем соединительной тканью или жиром.
Передается патология от в 70% случаев от матери, являющейся носителем мутировавшего гена. В оставшихся 30% мутации возникают в яйцеклетках женщины.
Симптомы миопатии Дюшена
Первые признаки болезни проявляются у больного ребенка в возрасте 1-5 лет, уже на втором году жизни можно заметить некоторое отставание моторного развития. Отмечается удлинение сроков сидения, вставания, хождения, обращает внимание неуклюжесть ребенка во время двигательной активности.
На 3-4 годах жизни начинает проявляться мышечная слабость. Первые ее признаки заключаются в аномальной утомляемости при длительных нагрузках. По мере прогрессирования патологии развивается «утиная походка».
Выраженная атрофия мышц начинается с тазового пояса и бедер с дальнейшим распространением на выше расположенные мышцы спины и плечелопаточного пояса, шеи.
Описаны следующие характерные признаки миопатии Дюшена:
- симптом Говерса — при переходе в вертикальное положение из сидячего ребенок опирается на свое тело, как бы взбираясь по нему;
- «осиная» талия и «крыловидные» лопатки — следствие выраженной атрофии мышц;
- резкое ослабление, а затем и полное исчезновение сухожильных коленных и плечевых рефлексов;
- искривление позвоночника и грудной клетки;
- деформация стоп.
У детей с миопатией Дюшена часто отмечается олигофрения, аутизм, СДГВ, синдром Иценко-Кушинга. Чаще всего к семилетнему возрасту большинство пациентов испытывают выраженные затруднения с двигательной активностью. к 12 годами почти 100% больных не могут самостоятельно передвигаться, а к 15 — совершать любые движения. В течение 1-5 лет после этого атрофируются дыхательные мышцы.
Диагностика миопатии Дюшена
Установить диагноз позволяют:
- тщательный сбор наследственного анамнеза;
- неврологическое обследование;
- исследование уровня креатинфосфокиназы в крови;
- электромиография.
Высокой степенью достоверности обладает генетический анализ, который позволяет выявить мутацию соответствующего гена. Однако в некоторых случаях мутацию определить не удается, в этом случае проводится биопсия мышц с иммуногистохимическим исследованием. Данный анализ позволяет выявить полное отсутствие белка дистрофина в мышцах.
Лечение и прогноз
Пока вылечить это заболевание невозможно, проводится симптоматическое и паллиативное лечение. Больным назначаются анаболические стероиды (деканоат, ретаболил), средства, улучшающие нервно-мышечную передачу (прозерин). С целью продления периода двигательной активности проводится ЛФК и физиотерапевтическое лечение.
В поздний период при нарушении функции дыхательных мышц назначается сначала вспомогательная вентиляция легких, а затем полная искусственная вентиляция. Большинство пациентов не доживают до 20 лет, но современные технологии позволяют значительно пролонгировать им жизнь.
Еще об одном генетическом заболевании- несовершенном остеогенезе - можно прочитать в этой статье.
Вконтакте
Google+
Одноклассники
boleznivse.ru
Мышечная дистрофия Дюшенна: симптомы и лечение
Мышечная дистрофия Дюшенна – это генетическое заболевание, связанное с нарушением строения мышечных волокон. Мышечные волокна при этой болезни, в конце концов, распадаются, и теряется способность к передвижению. Мышечная дистрофия Дюшенна передается сцеплено с полом, болеют лица мужского пола. Проявляет себя уже в детском возрасте. Помимо мышечных нарушений, заболевание приводит к скелетным деформациям, может сопровождаться дыхательной и сердечной недостаточностью, умственными и эндокринными нарушениями. Радикального способа лечения, позволяющего искоренить болезнь, пока нет. Все существующие меры являются лишь симптоматическими. Довольно редко больным удается пережить рубеж 30 лет. Эта статья посвящена причинам, симптомам, диагностике и лечению мышечной дистрофии Дюшенна.
Заболевание впервые описано в 1861 году (по другим данным – 1868 году) французским невропатологом и носит его имя. Встречается не так уж и редко: 1 случай на 3500 новорожденных детей. Из всех известных медицине мышечных дистрофий является наиболее распространенной.
Причина заболевания
В основе мышечной дистрофии Дюшенна лежит генетический дефект половой Х хромосомы.
Один из участков Х хромосомы содержит ген, кодирующий производство в организме особого мышечного белка под названием дистрофин. Белок дистрофин составляет основу мышечных волокон (миофибрилл) на микроскопическом уровне. Функция дистрофина заключается в поддержании клеточного скелета, в обеспечении способности миофибрилл к многократным актам сокращения и расслабления. При мышечной дистрофии Дюшенна этот белок либо отсутствует вообще, либо синтезируется дефектным. Уровень нормального дистрофина не превышает 3%. Это приводит к разрушению мышечных волокон. Мышцы перерождаются и заменяются жировой и соединительной тканью. Естественно, что при этом утрачивается двигательный компонент человеческой деятельности.
Заболевание наследуется по рецессивному типу, сцепленному с Х хромосомой. Что это означает? Поскольку все гены человека парные, то есть дублируют друг друга, то для того, чтобы появились патологические изменения в организме при наследственном заболевании, необходимо, чтобы генетический дефект возник в одной хромосоме или аналогичных участках обеих хромосом. Если заболевание возникает только при мутациях в обеих хромосомах, то такой тип наследования называют рецессивным. Когда же генетическая аномалия выявляется только в одной хромосоме, но болезнь все равно развивается, такой тип наследования называют доминантным. Рецессивный тип возможен только при одновременном поражении идентичных хромосом. Если вторая хромосома будет «здорова», то заболевание не возникнет. Именно поэтому мышечная дистрофия Дюшенна является уделом лиц мужского пола, потому что они имеют в генетическом наборе одну Х хромосому, а вторую (парную) – У. Если мальчику попадается «поломанная» Х хромосома, то у него обязательно возникает болезнь, потому что здоровой хромосомы у него просто нет. Для того, чтобы мышечная дистрофия Дюшенна возникла у девочки, необходимо совпадение в ее генотипе двух патологических Х хромосом, что практически маловероятно (в таком случае папа девочки должен быть болен, а у мамы в генетическом наборе должна содержаться дефектная Х хромосома). Девочки выступают лишь носителями заболевания и передают его своим сыновьям. Конечно, часть случаев заболевания не является результатом передачи по наследству, а возникает спорадически. Это означает появление мутации в генетическом наборе ребенка спонтанно. Вновь появившаяся мутация может быть передана по наследству (при условии сохранения способности к размножению).
Симптомы заболевания
Мышечная дистрофия Дюшенна всегда заявляет о себе до 5-летнего возраста. Наиболее часто первые симптомы возникают еще до 3-х лет. Все патологические проявления заболевания можно разделить на несколько групп (в зависимости от характера изменений):
- поражение скелетной мускулатуры;
- деформации скелета;
- поражение сердечной мышцы;
- умственные нарушения;
- эндокринные расстройства.
Поражение скелетных мышц
Поражение мышечной ткани является основным проявлением заболевания. Оно становится причиной генерализованной мышечной слабости. Начальные симптомы подкрадываются незаметно.
Рождаются дети без особых отклонений. Однако их двигательное развитие отстает в темпах по сравнению со сверстниками. Такие дети менее активные и подвижные в двигательном плане. Пока ребенок совсем мал, это часто связывают с особенностями темперамента и не обращают внимания на начальные изменения.
Явные признаки возникают с началом ходьбы. Дети часто падают и передвигаются на пальцах (на носочках). Следует отметить, что эти нарушения интерпретируют не при первых шагах ребенка, потому что прямохождение вначале для всех детей сопряжено с падениями и неуклюжестью. Когда большинство ровесников уже вполне уверенно передвигается, мальчики с мышечной дистрофией Дюшенна упорно продолжают падать.
Когда ребенок научится разговаривать, он начинает жаловаться на слабость и быструю утомляемость, непереносимость физических нагрузок. Бег, лазанье, прыжки и другие любимые виды активной деятельности детей для ребенка с мышечной дистрофией Дюшенна не привлекательны.
Походка таких детей напоминает утиную: они как бы переваливаются с ноги на ногу.
Своеобразным проявлением заболевания является симптом Говерса. Он заключается в следующем: при попытке ребенка подняться с колен, корточек, с пола он пользуется руками, чтобы помочь слабым мышцам ног. Для этого он опирается руками на себя, «взбираясь по лесенке, по самому себе».
Мышечная дистрофия Дюшенна имеет восходящий тип мышечной слабости. Это означает, что сначала слабость проявляет себя в ногах, затем распространяется на таз и туловище, потом на плечи, шею и, в конце концов, на руки, дыхательную мускулатуру и голову.
Несмотря на то, что при этом заболевании мышечные волокна подвергаются разрушению и развитию атрофии, внешне некоторые мышцы могут выглядеть вполне нормальными или даже накаченными. Развивается так называемая псевдогипертрофия мышц. Чаще всего этот процесс заметен в икроножных, ягодичных и дельтовидных мышцах, мышцах языка. Место распавшихся мышечных волокон занимает жировая ткань, именно поэтому создается эффект хорошей развитости мускулатуры, что на проверку оказывается совсем не так.
Атрофический процесс в мышцах всегда симметричный. Восходящее направление процесса приводит к возникновению «осиной» талии, «крыловидных» лопаток (лопатки отстают от туловища, словно крылья), симптома «свободных надплечий» (когда голова как бы проваливается в плечи при попытке поднять ребенка под мышки). Лицо гипомимично, губы могут утолщаться (замена мышц жировой и соединительной тканью). Псевдогипертрофия языка становится причиной речевых расстройств.
Разрушение мышц сопровождается развитием мышечных контрактур и укорочением сухожилий (хорошо заметно на примере ахиллового сухожилия).
Сухожильные рефлексы (коленный, ахиллов, с бицепса, с трицепса и так далее) постепенно снижаются. Мышцы на ощупь плотные, но безболезненные. Мышечный тонус обычно снижается.
Постепенное прогрессирование мышечной слабости приводит к тому, что к 10-12 годам многие дети утрачивают способность самостоятельно передвигаться и нуждаются в инвалидном кресле. Способность стоять сохраняется, в среднем, до 16 лет.
Отдельно следует сказать о вовлечении в патологический процесс дыхательной мускулатуры. Это наблюдается после подросткового периода. Слабость диафрагмы и других мышц, участвующих в акте дыхания, приводит к постепенному снижению жизненной емкости легких и объемов вентиляции. По ночам это особенно заметно (появляются приступы удушья), поэтому у детей могут возникать страхи перед сном. Формируется дыхательная недостаточность, которая усугубляет течение интеркуррентных инфекций.
Деформации скелета
Это сопутствующие мышечным изменениям симптомы. У детей постепенно формируются усиление поясничного изгиба (лордоза), искривление грудного отдела позвоночника в сторону (сколиоз) и сутулость (кифоз), меняется форма стопы. Со временем развивается диффузный остеопороз. Эти симптомы еще больше способствуют ухудшению двигательных нарушений.
Поражение сердечной мышцы
Является обязательным симптомом мышечной дистрофии Дюшенна. У больных развивается кардиомиопатия (гипертрофическая или дилатационная). Клинически это проявляет себя нарушениями сердечного ритма, перепадами артериального давления. Границы сердца увеличиваются, однако такое большое сердце имеет малые функциональные возможности. В конце концов, формируется сердечная недостаточность. Сочетание выраженной сердечной недостаточности с дыхательными расстройствами на фоне присоединившейся инфекции может быть причиной смертельного исхода у больных с мышечной дистрофией Дюшенна.
Умственные нарушения
Это не обязательный, но возможный признак заболевания. Он связан с дефицитом особой формы дистрофина – аподистрофином, содержащимся в головном мозге. Нарушения интеллекта варьируются от незначительных до степени идиотии. При этом выраженность умственных нарушений никоим образом не связана со степенью мышечных расстройств. Социальная дезадаптация из-за невозможности свободно передвигаться и посещать детские учреждения (садики, школы) способствует усугублению когнитивных расстройств.
Эндокринные расстройства
Встречаются у 30-50% больных. Могут быть довольно разнообразными, но чаще всего это ожирение с преимущественным отложением жира в области молочных желез, бедер, ягодиц, плечевого пояса, недоразвитие (или нарушение функции) половых органов. Больные часто имеют низкий рост.
Мышечная дистрофия Дюшенна неуклонно прогрессирует. К 15-20 годам почти все больные не в состоянии себя обслуживать самостоятельно ввиду обездвиженности. В конце концов, присоединяются бактериальные инфекции (органов дыхания и мочевыделительной сферы, инфицированные пролежни при недостаточном уходе), которые на фоне сердечной и дыхательной недостаточности приводят к смертельному исходу. Мало кто из больных переживает рубеж в 30 лет.
Диагностика
Диагностика мышечной дистрофии Дюшенна основывается на нескольких видах исследований, основным из которых является генетический тест (ДНК-диагностика).
Только обнаружение дефекта Х хромосомы в том участке, который отвечает за синтез дистрофина, достоверно подтверждает диагноз. До проведения такого анализа диагноз является предварительным.
Из других методов исследования могут применяться:
- определение активности креатинфосфокиназы (КФК). Этот фермент отражает гибель мышечных волокон. Его концентрация при мышечной дистрофии Дюшенна превышает норму в десятки и сотни раз до 5-летнего возраста. Позже уровень фермента постепенно снижается, потому что часть мышечных волокон уже необратимо разрушена;
- электромиография. Этот метод позволяет подтвердить тот факт, что в основе заболевания лежат первичные изменения мышц, а нервные проводники при этом совершенно интактны;
- биопсия мышц. С ее помощью определяют содержание белка дистрофина в мышце. Однако в связи с усовершенствованием генетической диагностики в последние десятилетия эта травматичная процедура отошла на второй план;
- дыхательные пробы (исследование жизненной емкости легких), ЭКГ, УЗИ сердца. Эти методы не используются для установления диагноза, но необходимы для выявления патологических изменений со стороны дыхательной и сердечно-сосудистой систем для того, чтобы скорригировать имеющиеся нарушения.
Выявление больного ребенка в семье означает, что в генотипе матери имеется патологическая Х хромосома. В редких случаях мать может быть здоровой, если мутация возникла у ребенка случайно. Наличие дефектной Х хромосомы несет в себе риск для последующих беременностей. Поэтому такие семьи должен консультировать генетик. При наступлении повторных беременностей родителям предлагают пренатальную диагностику, то есть исследование генотипа еще не родившегося ребенка с целью исключения наследственных заболеваний, в том числе мышечной дистрофии Дюшенна.
Для исследования понадобятся клетки плода, которые получают с помощью различных процедур на разных сроках беременности (например, биопсия хориона, амниоцентез и другие). И хотя эти медицинские манипуляции несут в себе определенный риск для беременности, они позволяют точно ответить на вопрос: имеется ли у плода генетическое заболевание.
Лечение
Мышечная дистрофия Дюшенна является в настоящее время неизлечимым заболеванием. Можно помочь ребенку (взрослому) продлить время двигательной активности, с помощью различных способов поддерживая мышечную силу, компенсируя изменения со стороны сердечно-сосудистой и дыхательной систем.
Несмотря на это, прогнозы ученых в отношении полного излечения от этого заболевания довольно оптимистичны, поскольку уже сделаны первые шаги в этом направлении.
В настоящее время, для лечения мышечной дистрофии Дюшенна из медикаментозных препаратов используют:
- стероиды (при регулярном применении они позволяют уменьшить мышечную слабость);
- β-2-адреномиметики (также временно придают силу мышцам, но не замедляют прогрессирование заболевания).
Применение β-2-адреномиметиков (Альбутерол, Формотерол) не имеет статистически достоверного признания, поскольку существует небольшой опыт их использования при данной патологии. Контроль изменений состояния здоровья у группы пациентов, применявших эти препараты, проводился в течение одного года. Поэтому утверждать, что они работают более длительное время, нет возможности.
Основой лечения сегодня являются стероиды. Считается, что их использование позволяет какое-то время сохранять мышечную силу, то есть они могут затормозить прогрессирование болезни. Кроме того, доказано, что стероиды уменьшают риск возникновения сколиоза при мышечной дистрофии Дюшенна. Но все же возможности этих препаратов ограничены, и заболевание будет неуклонно прогрессировать.
Когда же начинают лечение гормонами? Считается, что оптимальным временем для начала терапии является такая фаза заболевания, когда двигательные навыки не улучшаются, но еще не ухудшаются. Обычно это бывает в возрасте 4-6 лет. Наиболее часто используются такие препараты, как Преднизолон и Дефлазакорт. Дозы назначаются индивидуально. Препараты используются, пока есть видимый клинический эффект. Когда же наступает фаза прогрессирования заболевания, то необходимость применения стероидов отпадает, и их постепенно (!) отменяют.
Из медикаментозных препаратов также при мышечной дистрофии Дюшенна применяют сердечные средства (антиаритмические, метаболические, ингибиторы ангиотензинпревращающего фермента). Они позволяют бороться с кардиологическими аспектами заболевания.
Из немедикаментозных методов лечения значимую роль играют физиотерапия и ортопедическая помощь. Физиотерапевтические методики позволяют дольше, чем без их применения, сохранять гибкость и подвижность суставов, поддерживают мышечную силу. Доказано, что умеренная физическая активность благоприятно влияет на течение заболевания, а вот бездействие и постельный режим наоборот способствуют еще более быстрому прогрессированию заболевания. Поэтому необходимо как можно дольше поддерживать посильную физическую деятельность, даже после того, как больной «пересел» в инвалидное кресло. Показаны регулярные курсы массажа. На самочувствие больного положительно влияет плавание.
Ортопедические приспособления позволяют значительно облегчить жизнь больного. Их перечень довольно широк и разнообразен: это и различного рода вертикализаторы (помогают сохранять положение стоя), и приспособления для самостоятельного вставания, и инвалидные кресла-коляски с электрическим приводом, и специальные шины для устранения контрактур в голени (используются даже ночью), и корсеты для позвоночника, и длинные шины для ног (колено-голеностопные ортезы), и многое другое.
Когда заболевание поражает и дыхательные мышцы, и самостоятельное дыхание становится неэффективным, то возможно применение аппаратов искусственной вентиляции легких различной модификации.
И все же даже использование всех этих мер в комплексе не позволяет побороть заболевание. На сегодняшний день, есть ряд перспективных направлений исследований, которые, возможно, станут прорывом в лечении мышечной дистрофии Дюшенна. К наиболее распространенным среди них относят:
- генную терапию (введение «правильного» гена с помощью вирусных частиц, доставку генетических конструкций в составе липосом, олигопептидов, полимерных носителей и другие);
- регенерацию мышечных волокон с помощью стволовых клеток;
- трансплантацию миогенных клеток, которые способны к синтезу нормального дистрофина;
- пропуск экзонов (с помощью антисмысловых олигорибонуклеотидов) в качестве попытки замедлить прогрессирование заболевания и смягчить его течение;
- замену дистрофина на другой белок атрофин, ген которого расшифрован. Методика опробована на мышах и дала положительные результаты.
Каждая из новых разработок несет в себе надежду для больных с мышечной дистрофией Дюшенна на полное выздоровление.
Таким образом, мышечная дистрофия Дюшенна – это генетическая проблема лиц мужского пола. Болезнь характеризуется прогрессирующей мышечной слабостью из-за разрушения мышечных волокон. В настоящее время является неизлечимым заболеванием, однако многие ученые мира трудятся над созданием радикального способа борьбы с ним.
Мультипликационный фильм «Мышечная дистрофия Дюшенна», англ. озвучка, субтитры на русском языке:
doctor-neurologist.ru