Лечение суставов - артроз, артрит, остеохондроз и многое другое
Амиотрофия верднига гоффмана
Спинальная амиотрофия Верднига-Гоффмана: причины, симптомы, лечение
Спинальная амиотрофия Верднига-Гоффмана (острая злокачественная инфантильная спинальная амиотрофия Верднига-Гоффмана, спинальная амиотрофия I типа) – это наследственное заболевание нервной системы, характеризующееся развитием мышечной слабости практически во всех мышечных структурах организма. Приводит к нарушению способности к сидению, передвижению и самообслуживанию. Эффективных средств лечения заболевания не существует. Дородовая диагностика помогает избежать рождения больного ребенка в семье. Из этой статьи Вы сможете узнать о том, как наследуется это заболевание, чем проявляется и чем же можно помочь таким больным.
Болезнь носит название двух ученых, которые впервые описали ее. В конце XIX века Верднигом и Гоффманом была доказана морфологическая сущность заболевания. Они предполагали единственную подобную форму заболевания. Однако в XX веке Кукельбергом и Веландером была описана иная клиническая форма спинальной амиотрофии, которая имела ту же генетическую причину, что и спинальная амиотрофия Верднига-Гоффмана. На сегодняшний день под понятием спинальной амиотрофии объединяют несколько клинически отличающихся форм болезни. Но все они связаны с одним и тем же наследственным дефектом.
Причины спинальной амиотрофии
Заболевание наследственное. В основе лежит генетическая мутация в 5-й хромосоме человека. Мутации подвергается ген, отвечающий за производство белка SMN. Синтез этого белка обеспечивает нормальное развитие двигательных нейронов. В случае развития мутации двигательные нейроны разрушаются или оказываются недоразвитыми, а значит, передача импульса с нервного волокна к мышце оказывается невозможной. Мышца не работает. Следовательно, все движения, связанные с неработающей мышцей, не выполняются.
Неправильный ген имеет аутосомно-рецессивный тип наследования. Это означает следующее: для того, чтобы развилась спинальная амиотрофия, необходимо совпадение двух мутантных генов от матери и от отца. То есть мать и отец ребенка должны быть носителями патологического гена, но при этом они не больны ввиду одновременного наличия у них здорового доминантного (преобладающего) гена (гены у каждого человека парные). Если мать и отец являются носителями патологического гена, то риск рождения больного ребенка составляет 25%. Подсчитано, что приблизительно каждый 50-й человек на планете является носителем мутантного гена.
Симптомы
На сегодняшний день известно 4 формы спинальной амиотрофии. Все они отличаются по сроку возникновения заболевания, некоторым симптомам и длительности жизни. Общим для всех форм является отсутствие чувствительных и умственных нарушений. Функции тазовых органов никогда не страдают. Все симптомы связаны только с поражением двигательной сферы.
Спинальная амиотрофия I типа
 Дебют болезни в возрасте до 6 месяцев имеет крайне неблагоприятный прогноз.
Дебют болезни в возрасте до 6 месяцев имеет крайне неблагоприятный прогноз.Возможны нарушения сосания и глотания, затрудненные движения языка. На самом языке могут быть видны фасцикуляции (непроизвольные мышечные сокращения, «волны» бегают по языку), а сам он выглядит атрофированным. Крик ребенка вялый и слабый. Если снижается глоточный рефлекс, то возникают проблемы с кормлением, в результате чего пища попадает в дыхательные пути. А это вызывает аспирационную пневмонию, от которой ребенок может погибнуть.
Поражение диафрагмы и межреберных мышц проявляется нарушением акта дыхания. Вначале этот процесс компенсирован, но постепенно дыхательная недостаточность усугубляется.
Характерно то, что мимические мышцы лица и мышцы, отвечающие за движения глаз, не поражаются.
Такие дети отстают в моторном развитии: они не держат голову, не переворачиваются, не тянутся за предметом, не сидят. Если какие-то двигательные навыки и смогли реализоваться до начала заболевания, то они будут утрачены.
Кроме двигательных нарушений заболевание характеризуется деформацией грудной клетки.
Если признаки заболевания видны сразу после рождения, то такие дети часто погибают в течение первых 6 месяцев жизни. Если признаки появляются после 3 месяцев, то срок жизни несколько больше – около 2-3 лет. Неизбежно присоединяется инфекция вследствие дыхательных нарушений, от которой такие детки и умирают.
Спинальная амиотрофия может сочетаться с врожденными пороками развития: олигофренией, маленьким черепом, пороками сердца, врожденными переломами, гемангиомами, косолапостью, неопущением яичек.
Спинальная амиотрофия II типа
Эта форма заболевания возникает в промежутке между первыми 6 месяцами и 2 годами жизни. До этого у ребенка не выявляют никаких нарушений. Он вовремя начинает держать голову, переворачиваться и сидеть, а иногда и ходить. А затем постепенно появляется мышечная слабость. Обычно все начинается с мышц бедер. Потихоньку становится невозможной ходьба, снижаются и утрачиваются сухожильные рефлексы. Мышечная слабость прогрессирует медленно. Вовлекаются все конечности. Развивается атрофия мышц. Процесс может захватывать и дыхательную мускулатуру. Также, как и при I-м типе спинальной амиотрофии, не поражаются мимические мышцы и мышцы глаза. Возможно дрожание кистей, подергивания на языке и конечностях. Слабость мышц шеи проявляется свисанием головы.
Весьма характерными являются костно-суставные деформации: сколиоз, воронкообразная грудная клетка, вывих тазобедренного сустава.
Эта форма имеет более доброкачественное течение, чем спинальная амиотрофия I типа, но у большинства больных к подростковому периоду имеются дыхательные нарушения. Плохая экскурсия грудной клетки способствует присоединению инфекций, от которых ребенок может погибнуть.
Спинальная амиотрофия III типа
Эта форма описана Кукельбергом и Веландером. Считается юношеской спинальной амиотрофией. Начало заболевания между 2 и 15 годами.
Первым симптомом всегда становится неустойчивая ходьба из-за нарастающей слабости в ногах. Тонус в ногах снижается, развивается мышечная атрофия (мышцы истончаются), но это не всегда заметно из-за хорошо развитого к этому возрасту слоя подкожной жировой клетчатки. Дети спотыкаются, падают, неловко передвигаются. Постепенно движения в ногах становятся невозможными, и больной перестает ходить.
Исподволь заболевание захватывает и верхние конечности, кисти рук поражаются позже. При этой форме развивается слабость мимических мышц, но движения глаз сохраняются в полном объеме. Отсутствуют рефлексы с тех мышечных групп, которые уже вовлечены в процесс.
Также характерны деформации скелета: воронкообразная грудная клетка, контрактуры суставов.
Эта форма заболевания при проведении поддерживающей терапии позволяет больным жить до 40 лет.
Спинальная амиотрофия IV типа
Эта форма заболевания считается «взрослой», потому как проявляется после 35 лет. Также возникает слабость в мышцах ног, снижение рефлексов, атрофии мышц, что, в конце концов, приводит к полной утрате движений в ногах. При этом дыхательная мускулатура не вовлекается в процесс, нарушений дыхания не бывает. Продолжительность жизни при этой форме заболевания почти такая же, как и у здоровых людей. Течение наиболее доброкачественное по сравнению с другими формами.
Диагностика
При появлении симптомов, похожих на спинальную амиотрофию, проводят электронейромиографию (обнаруживают спонтанную активность в виде потенциалов фасцикуляций в покое и повышение средней амплитуды потенциалов действия двигательных единиц).
Окончательно вопрос о диагнозе решается после генетического исследования (ДНК-диагностика): находят мутацию гена в 5-й хромосоме.
В семьях, где были случаи подобных заболеваний, проводят пренатальную (дородовую) ДНК-диагностику у плода. При выявлении патологии решают вопрос о прерывании беременности.
Принципы лечения спинальной амиотрофии
К сожалению, это неизлечимое наследственное заболевание. На современном этапе проводятся исследования, которые, возможно, помогут регулировать синтез белка SMN, но результатов пока нет.
Облегчить состояние больным со спинальной амиотрофией помогают:
- периодический курсовый прием препаратов, улучшающих метаболизм нервной ткани и мышц (Церебролизин, Цитофлавин, Глутаминовая кислота, АТФ, Карнитина хлорид, Метионин, Калия оротат, Токоферола ацетат и др.);
- витамины группы В (Мильгамма, Нейровитан, Комбилипен);
- анаболические стероиды (Ретаболил, Неробол);
- средства, улучшающие нервно-мышечную проводимость (Прозерин, Нейромидин, Галантамин, Дибазол);
- курсы массажа и лечебной физкультуры;
- физиотерапия (электростимуляция мышц, углекисло-сульфидные ванны);
- методы ортопедической коррекции (при развитии контрактур суставов и деформации позвоночника).
Спинальная амиотрофия Верднига–Гоффмана, как и другие формы этого заболевания, является патологией, передающейся по наследству. Появление болезни у ребенка объясняется наличием мутантного гена и у матери, и у отца. Болезнь характеризуется, в основном, мышечной слабостью, которая становится причиной обездвиженности и дыхательных нарушений. Заболевание, на сегодняшний день, неизлечимо.
doctor-neurologist.ru
Спинальная амиотрофия Верднига-Гоффмана - причины, симптомы, диагностика, лечение
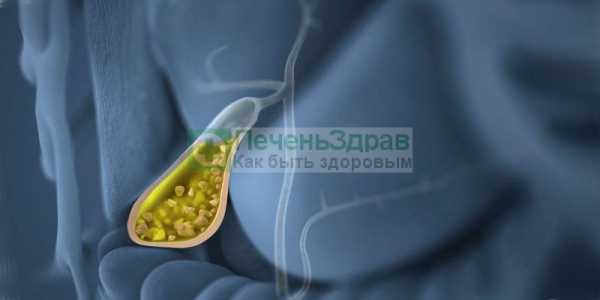
Это группа наследственно обусловленных заболеваний, главной чертой которых является поражение мотонейронов передних рогов спинного мозга, а также поражение корешков IX, X, XII черепно-мозговых нервов.
Для спинальной амиотрофии характерно нарушение иннервации мышц нижних конечностей, шеи, головы, дыхательной мускулатуры. Важными критериями постановки правильного диагноза является сохранение всех видов чувствительности, нормальное развитие мышц верхних конечностей и отсутствие у ребенка психических нарушений. Частота встречаемости заболевания составляет 7 человек на 100 000 новорожденных.
Причины амиотрофии Верднига-Гоффмана
Ген спинальной амиотрофии Верднига-Гоффмана (SMN) локализован на V хромосоме, он наследуется по аутосомно-рецессивному типу. Родители, хромосомы которых несут ген SMN, могут произвести на свет ребенка, больного спинальной амиотрофией с вероятностью 25%.
Патоморфологические изменения при амиотрофии Верднига-Гоффмана
При исследовании отмечается уменьшение спинного мозга в объеме. Атрофируются или полностью исчезают ганглиозные клетки. В передних корешках выявляют дегенерацию, демиелинизацию, склеротические изменения нервных волокон (пери-, эпи-, эндоневральный) с отложением жира. В мышцах скелета обнаруживаются атрофированные пучки, которые переплетаются с интактными волокнами, отмечается гиалиноз, разрастание соединительной ткани.
Классификация заболевания амиотрофии Верднига-Гоффмана
По времени возникновения и степени дистрофических изменений при спинальной амиотрофии Верднига – Гоффманна:
- Врожденная (появление симптомов заболевания первые 6 месяцев жизни);
- Ранняя детская (от 6 мес до 1.5 лет);
- Поздняя детская (старше 1.5 лет).
Симптомы амиотрофии Верднига-Гоффмана
Самой тяжелой является врожденная форма спинальной амиотрофии Верднига-Гоффмана. У детей уже на первых минутах жизни отмечаются вялые парезы. Выявляется мышечная слабость, сниженные рефлексы периода новорожденности или их отсутствие. Новорожденные слабо сосут грудь, у них отмечаются фасцикулярные подергивания языка, глотание затруднено. Эта форма заболевания сопровождается формированием костно-мышечных деформаций, в частности сколиотическими; воронкообразной или «куриной» грудью; контрактурами суставов. В очень редких случаях ребенок имеет способность держать голову, садится. Однако эти способности развиваются поздно, а далее регрессируют. Данному заболеванию часто сопутствуют врожденные аномалии, такие как гидроцефалия, дисплазия т/б суставов, плосковальгусные или плосковарусные деформации стоп, неопущение яичек в мошонку, гемангиомы и т. д. Дети умирают до 9 месяцев (реже до 2-х лет) от сердечно-сосудистой либо дыхательной недостаточности, причина которых – гипотония грудных мышц и мышц диафрагмы.
Ранняя детская форма спинальной амиотрофии Верднига-Гоффмана характеризуется манифестацией во втором полугодии. Больной ребенок своевременно начинает удерживать голову, сидит, иногда он может даже стоять или ходить. Далее после перенесенного алиментарного энтероколита происходит прогрессирование состояния: вялые парезы проявляются сначала на ногах, далее поднимаются на тело и верхние конечности. Вследствие диффузной мышечной атрофии отмечаются фасцикулярные подергивания языка, контрактуры, мелкий тремор кистей. Бульбарный синдром развивается значительно позже. Протекает ранняя детская форма спинальной амиотрофии Верднига-Гоффмана не так злокачественно, как первый вариант заболевания, тем не менее, летальный исход наступает к 12-15 годам. Поздняя форма заболевания проявляется в возрасте у дошкольников. На фоне мнимого благополучия, когда ребенок самостоятельно передвигается, прыгает, бегает, появляются скованность, движения становятся неловкими (походка «заводной куклы»), дети часто спотыкаются. Атрофия мышц скелета происходит постепенно и медленно: сначала вялые парезы наблюдаются в нижних отделах нижних конечностей, далее в процесс вовлекаются мышцы нижних отделов верхних конечностей, туловища. Мышечная атрофия может остаться незаметной, т. к. в этом возрасте хорошо развита подкожная жировая клетчатка. Постепенно происходит ослабление таких рефлексов, как глоточный и небный, снижаются безусловные рефлексы. Заболеванию сопутствуют деформации опорного аппарата, чаще всего это «куриная» грудь. Прогноз при адекватной и своевременной терапии более благоприятный относительно первых двух вариантов. Больные могут доживать до 30-40 лет. Однако способность самостоятельно передвигаться исчезает к 10-12 годам.
В литературе иногда можно встретить четвертую форму заболевания – взрослую, которая манифестирует в возрасте старше 35 лет. Это крайне редкая и наиболее благоприятная форма болезни, при которой происходит нарушение иннервации только мышечных групп нижних конечностей. У таких пациентов теряется способность к самостоятельному передвижению, но отсутствуют нарушения дыхания, глотания. Взрослая форма амиотрофии не сказывается на продолжительности жизни больных.
Диагностика АДС Верднига-Гоффманна
Диагноз подтверждается на основании клинической картины (раннее начало атрофических изменений, начало дегенеративных изменений в проксимальных группах мышц, гипотония мышц, подергивания мышц языка, отсутствие псевдогипертрофии), данных ЭНМГ (электронейромиографии), результата биопсии мышечных волокон, МРТ, генеалогического анализа (поиск генетических мутаций у родителей и ребенка). Заболевание имеет быстропрогрессирующее течение.
Дифференциальный диагноз амиотрофии Верднига-Гоффмана
1. С прочими заболеваниями, характеризующимися «синдромом вялого ребенка»; 2. Генетическими заболеваниями обмена веществ; 3. Амиотрофией Оппенгейма (в настоящее время некоторыми специалистами рассматривается как вариант спинальной амиотрофии Верднига-Гоффмана); 4. ДЦП; 5. Прогрессирующими мышечными дистрофиями (Дюшена и Эрба-Рота); 6. Амиотрофией Кугельберга-Веландера;
7. Свинцовой интоксикацией.
Лечение амиотрофии Верднига-Гоффмана
Спинальная амиотрофия Верднига-Гофмана на данный момент неизлечимое, постоянно прогрессирующее заболевание. Существует только симптоматическая терапия: препараты, которые действуют на обменные процессы нервной ткани (церебролизин; аминолон; энцефабол); ноотропы (луцетам, ноотропил); витамины группы В; массаж и ЛФК, специальная диета и т. д.
С генетическими мутациями при спинальной амиотрофии связано уменьшение выработки SMN-белка, что приводит к потере моторных нейронов. Задачей номер один современной фармакологии при этом заболевании является поиск препарата, способного увеличить уровень SMN-белка.
Профилактика амиотрофии Верднига-Гоффмана
Заключается в своевременной диагностике генетических нарушений у родителей, пренатальная ДНК-диагностика. При выявлении патологии у плода – решение вопроса о прерывании беременности.
medlibera.ru
medlibera.ru
Спинальная амиотрофия Верднига-Гоффмана — симптомы, лечение и фото патологии
Генетическое заболевание Верднига-Гофмана относится к группе спинальных амиотрофий, наследуется по аутосомно-рецессивному типу.
Спинальная мышечная атрофия (СМА) характеризуется врожденными или приобретенными дегенеративными изменениями в поперечнополосатых мышцах, симметричной мышечной слабостью туловища, конечностей, отсутствием или снижением сухожильных рефлексов при сохранении чувствительности.
Морфологические исследования выявляют патологию двигательных нейронов спинного мозга, «пучковую атрофию» в скелетных мышцах с характерным чередованием пораженных волокон и здоровых.
Отмечается нарушение проводящей функции нервных волокон, снижение сократительной способности мышц. Статистика
1 из 40-50 человек является носителем мутантного гена SMN. Проявляется патология с частотой 1 : 6 000 — 10 000 новорожденных.
Причины заболевания
Основной причиной спинальной амиотрофии Верднига Гоффмана является мутация гена SMN (от англ. survival motor neuron). Располагается ген выживания мотонейрона на 5 хромосоме, представлен двумя копиями:
- SMNt — теломерная копия, функционально активная;
- SMNc — центромерная копия гена, частично активная.
Продуктом этого гена является белок SMN, участвующий в образовании и регенерации РНК.
Нехватка белка вызывает патологии двигательного нейрона.
В 95% случаев болезни Верднига-Гофмана наблюдается делеция (выпадение) SMNt, что вызывает дефицит белка SMN. Копия SMNc лишь частично компенсирует отсутствие теломерной копии.
Количество копий SMNc составляет от 1 до 5. Чем больше число центромерных копий, тем полнее воспроизводится белок и менее выражена патология нейрона.
Кроме количества копий SMNc, тяжесть заболевания определяется длиной участка делеции и генными конверсиями еще 3 генов: NAIP, h5F5, GTF2h3. Участием дополнительных модифицирующих факторов объясняется клиническое разнообразие симптомов.
Формы спинальной амиотрофии Верднига Гофмана
Выделяю такие виды:
- ранняя детская или СМА 1 – признаки заболевания проявляются до 6 месячного возраста;
- поздняя форма или СМА 2 – симптомы появляются после 6 месяцев до 1 года.
Симптоматика заболевания
СМА 1 и СМА 2 имеет разные симптомы и признаки.
Форма спинальной амиотрофии Верднига СМА 1
Первые симптомы выявляют еще во время беременности по слабому шевелению плода.

Фото: спинальная амиотрофия Верднига Гофмана
С самого рождения у детей наблюдается дыхательная недостаточность, врожденная спинальная амиотрофия Верднига Гофмана отмечаются:
- низкий мышечный тонус, ребенок не держит голову, не может перевернуться;
- отсутствие рефлексов;
- нарушения сосания, глотания, подергивание языка, пальцев, слабый плач.
Малыш принимает характерную позу «лягушки» с согнутыми в суставах руками и ногами, лежа на животе. При СМА 1 нередко отмечают частичный паралич диафрагмы – синдром Кофферата.
Явление характеризуется затруднением дыхания, одышкой, цианозом.
На стороне паралича наблюдается выбухание грудной клетки, повышается риск пневмонии.
У младенцев наблюдаются деформации костной системы, выражающиеся в ограничении подвижности суставов, появлении сколиоза, изменении формы грудной клетки.
Первые месяцы жизни дети развиваются нормально: вовремя начинают держать головку, сидеть, стоять.
После 6 месяцев появляются первые симптомы, обычно после острой респираторной или пищевой инфекции.
В первую очередь страдают конечности, особенно ноги, снижаются сухожильные рефлексы.
Затем в процесс постепенно вовлекаются мышцы туловища и рук, межреберные мышцы, диафрагма, что вызывает деформацию грудной клетки. Изменяется походка, приобретая сходство с «заводной куклой».
Дети становятся неловкими, часто падают. Наблюдаются подергивания языка, дрожание пальцев.
Течение болезни
СМА 1 характеризуется злокачественным течением. Тяжелые расстройства дыхательной функции, сердечно-сосудистая недостаточность нередко приводят к смерти в первые месяцы жизни. До 5 лет доживают 12% больных.
СМА 2 также имеет тяжелый прогноз, хотя и протекает несколько мягче. Летальный исход отмечается в 14-15 лет.
При спинальной амиотрофии Вердника диагностика заключается в проведении генетического анализа, выявляя мутации или делецию гена SMN.
При обнаружении делеции теломерной копии SMNt диагноз считают подтвержденным.
В случае отсутствия делеции проводят дополнительные исследования:
- электонейромиографию;
- исследование нервной проводимости;
- тест на креатинкиназу;
- биопсию мышц и нервной ткани.
При нормальных показателях фермента креатинкиназы проводят подсчет копий SMNc. В случае единственной копии идентифицируют точечную мутацию, принимая окончательное решение.
Дифференциальная диагностика
Похожие симптомы наблюдаются при врожденной миопатии – нарушении тонуса мышц.
Полностью исключить мышечную гипотонию позволяют результаты биопсии.
Определенное сходство с заболеванием Верднига-Гофмана имеет острый полиомиелит. Он начинается бурно, с резкого подъема температуры, несимметричных множественных параличей.
Несколько дней длится острый период, затем процесс переходит в восстановительную стадию.
Гликогенозы и врожденные миопатии также характеризуются сниженным мышечным тонусом. Изменения вызываются, в отличие от спинальной мышечной амиотрофии, нарушением обмена веществ, карциномой, гормональным дисбалансом. Следует исключить также болезнь Гоше, синдром Дауна, ботулизм.
Лечебные методики
Лечение спинальной амиотрофии симптоматическое и направлено на стабилизацию состояния пациента.
Назначают лекарственные средства:
- улучшающие метаболизм – церебролизин, липоцеребин, аминалон;

- влияющие на трофику мышечной ткани – оротат калия, глютаминовая кислота, метионин, токоферола ацетат;
- способствующие нервно-мышечной проводимости – прозерин, галантамин, дибазол;
- стимулирующие кровообращение в капиллярах – компламин, никотиновая кислота;
- поддерживающие жизнеспособность двигательных нейронов – вальпроевую кислоту, рилузол, L-карнитин.
Больным предписывают ортопедические процедуры в сочетании с теплыми ваннами, показаны лечебная гимнастика, мягкий массаж, оксигенотерапия, сульфидные ванны.
Виды спинальных амиотрофий
Условно различают проксимальные и дистальные формы СМА. 80% всех видов спинальных амиотрофий относятся к проксимальной форме.
К ним относятся, кроме заболевания Верднига-Гофмана:
- СМА 3 или болезнь Кульдберга-Веландер — заболевают в возрасте от 2 лет до 20, первыми страдают мышцы таза. Отмечается тремор кистей, лордоз.
- Летальная X-сцепленная форма — описана в 1994 году Baumbach, наследуется по рецессивному признаку, наблюдаются преимущественно поражения мышц таза и плечевого пояса.
- Инфантильная дегенерация — нарушаются рефлексы сосания, глотания, дыхание. Смерть может последовать в возрасте до 5 месяцев.
- СПА Рюкю — ген сцепливания не выявлен, наблюдается отсутствие рефлексов, мышечная слабость конечностей после рождения.
В эту группу входит также болезнь Нормана, СМА с врожденным артрогрипозом, СМА с врожденными переломами.
К дистальным спинальным амиотрофиям относится прогрессирующий детский паралич Фацио-Лонде, болезнь Брауна-Виалетта-ван Лэре, СМА с параличом диафрагмы, эпилепсией и глазодвигательными нарушениями.
Читайте ещё
neurodoc.ru
Болезнь Верднига-Гоффмана (Спинальная амиотрофия)
Спинальная амиотрофия представляет собой прогрессирующее нервно-мышечное заболевание, имеющее несколько форм течения. Болезнь передается по наследству.
При этой патологии страдает проксимальная поперечнополосатая мускулатура – преимущественно, нижних конечностей, туловища и шеи. Чувствительность в пораженных областях сохранена, интеллектуальное и психическое развитие не нарушены.
Самые тяжелые формы болезни с неблагоприятным прогнозом отмечаются при манифестировании болезни в детском возрасте.
Общие сведения
Свое название в виде «синдрома Верднига-Гоффмана» спинальная мышечная атрофия (СМА) получила благодаря ученым-неврологам G. Werdnig и J. Hoffmann. Они описали основные проявления болезни у детей еще в конце XIX столетия. В связи с особенностями русской транскрипции фамилии второго автора, можно встретить и другие названия этого синдрома в отечественной литературе: Верднига-Гофмана или -Хоффмана.
В середине XX столетия Е. Kugelberg и L. Welander описали поздние формы СМА, которые имеют такое же происхождение, что и детские, но их течение более доброкачественное. Поэтому иногда поздние формы спинальной амиотрофии (третий и четвертый тип) называют синдромом Кугельберга-Вилландера.
Причины появления СМА
Болезнь развивается в результате поражения двигательных (моторных) нейронов, находящихся в передних рогах спинного мозга, вплоть до полного их исчезновения. Последствием этого является нарушение иннервации проксимальных мышечных волокон, что приводит к прогрессирующей их слабости, развитию атрофии и невозможности совершать движения.
Причиной такого разрушения является дефицит специального белка, за синтез которого отвечает ген SMN (survival motor neuron gene). Локализация этого гена находится в пятой хромосоме. Генетически обусловленная мутация гена SMN, собственно, и вызывает развитие этой патологии.
Болезнь Верднига-Гофмана относится к аутосомно-рецессивному типу наследования. Это значит, что аномальный ген должен присутствовать у обоих родителей, которые при этом могут не иметь внешних признаков заболевания. При этом риск рождения больного ребенка у такой пары составляет 25%. В остальных случаях рождаются абсолютно здоровые дети.
Статистика и факты о СМА:
- Один ребенок из 6000–10000 новорожденных будет иметь признаки спинальной амиотрофии.
- Рецессивный ген, «ответственный» за развитие заболевания, выявляется примерно у каждого 50-го человека.
- Спинальная амиотрофия является одной из распространенных генетических нарушений, хотя встречается относительно редко.
- Проявления болезни могут манифестировать в любом возрасте.
- Половина заболевших детей не доживают до двухлетнего возраста.
Помимо этой патологии, существуют также виды спинальной амиотрофии с аутосомно-доминантным или сцепленным с Х-хромосомой типом наследования. Однако, для них характерны мутации других генов и, соответственно, иные признаки.
Типы болезни
В классификации синдрома Верднига-Гоффмана выделяют четыре типа течения болезни. В основу такого подразделения положены следующие критерии:
- Возраст начала заболевания (манифестации).
- Тяжесть течения.
- Продолжительность жизни.
По мнению многих исследователей, именно к синдрому Верднига-Гофмана относятся только первый и второй типы спинальной амиотрофии. Но, учитывая отсутствие общепринятой классификации, мы будем рассматривать проявления всех типов этой болезни.
Отмечено, что «толчком» к началу дебюта всех типов СМА нередко служит перенесенное инфекционное заболевание, травма, проведенная вакцинация или другой внешний фактор.
Первый тип
Такая форма синдрома Верднига-Гофмана манифестирует в возрасте до 6 месяцев жизни ребенка. Первый тип СМА отличается самым злокачественным и быстропрогрессирующим течением.
Иногда заболевание начинает развиваться еще во внутриутробном периоде. В таком случае диагностируется низкая активность плода, вялые и редкие шевеления. Если дебют спинальной амиотрофии произошел позже, то такие дети могут нормально держать голову и даже пытаться переворачиваться. Правда, эти умения быстро утрачиваются.
Общие симптомы для первого типа СМА:
- Быстрое развитие общей мышечной гипотонии. Для ребенка характерна поза «лягушки»: безвольное отведение ног и рук с согнутыми коленными и локтевыми суставами. Сухожильные рефлексы отсутствуют.
- Нарушены сосательные и глотательные функции, затруднены движения языка.
- Крик таких детей слабый.
- Поражение межреберных и диафрагмальных мышц вызывают проблемы с актом дыхания с развитием дыхательной недостаточности.
- Часто развиваются деформации грудной клетки.
Нередко врожденная спинальная амиотрофия первого типа сочетается с аномалиями развития. Наиболее часто выявляется микроцефалия, косолапость, крипторхизм у мальчиков, миопатии, врожденные переломы и т. д.
Часто смерть таких детей наступает от осложненной пневмонии (аспирационной или инфекционной) и сопутствующей прогрессирующей дыхательной недостаточности.
Дети с врожденной формой СМА первого типа редко доживают до 12 месяцев жизни. При более поздней манифестации этой формы заболевания продолжительность жизни составляет обычно два-три года.
Второй тип
Признаки синдрома Верднига-Гофмана в этом случае впервые появляются в возрасте 6–18 месяцев жизни ребенка. До начала дебюта заболевания ребенок ничем не отличается от здоровых сверстников. Такие дети нормально держат голову, умеют ползать, сидеть и даже стоять. Однако, в большинстве случаев, научиться ходить они так и не успевают.
Характерные симптомы второго типа СМА:
- Быстрый регресс полученных навыков – ползания, сидения, стояния и т. д. Ребенок перестает стоять, затем – сидеть, ползать и переворачиваться.
- Типично распространение мышечной слабости начиная с нижних конечностей вверх, с развитием вялых парезов.
- Верхние конечности обычно поражаются позже.
- Вследствие поражения мышц шеи ребенок плохо удерживает голову в вертикальном положении.
- Характерными являются миофасцикуляции (непроизвольные подергивания мышечных пучков). Чаще они возникают в кистях, языке, мышцах плечевого и тазового поясов.
- Тремор (дрожание) рук.
- Нередко возникает контрактура (неподвижность) суставов и позвоночного столба, обусловленная мышечной гипотонией и атрофией.
- Позже возникают признаки бульбарного паралича. При этом у таких детей нарушается произношение звуков вплоть до полной афонии. Происходят расстройства акта глотания: больные поперхиваются даже жидкой пищей или не могут проглотить еду и жидкость.
Продолжительность жизни больных со вторым типом СМА составляет приблизительно 10–14 лет. Смерть обычно наступает вследствие глубоких расстройств ритма дыхания и сердечной деятельности.
Причиной летального исхода достаточно часто также является пневмония.
Третий и четвертый тип
Спинальная амиотрофия в этом случае манифестирует в достаточно широком возрастном диапазоне: от 1,5–2 до 20 и старше лет. При этом дети уже овладевают основными двигательными навыками и умеют сидеть, стоять, ходить и бегать. Заболевание развивается постепенно и начинается часто незаметно.
Для такого типа спинальной амиотрофии характерно медленное развитие патологических симптомов, которое занимает иногда достаточно длительное время. Способность больных к самостоятельному передвижению и самообслуживанию сохраняется 8–10 и более лет от дебюта заболевания.
Признаки поздней формы СМА:
- Мышечная слабость начинает развиваться с проксимальных отделов нижних конечностей. Такие дети быстро устают при ходьбе, спотыкаются, плохо бегают.
- С прогрессированием болезни возникает характерная походка, со сгибанием ног в коленных суставах («заводной куклы»).
- Со временем развиваются вялые парезы и параличи нижних конечностей, что обуславливает неспособность больного к самостоятельному передвижению.
- Постепенно поражаются мышцы туловища, шеи и верхних конечностей.
- При вовлечении в патологический процесс бульбарного отдела возникают расстройства речи и функции глотания.
- Мышечная атрофия достаточно долго не выявляется визуально, вследствие хорошо выраженного подкожно-жирового слоя. Нарастание ее интенсивности сопровождается угнетением сухожильных рефлексов и формированием суставных контрактур.
- Также могут формироваться различные деформации костей и суставов. Помимо суставных контрактур, наиболее часто встречаются гиперлордоз поясничного отдела позвоночника и воронкообразный вид грудной клетки.
- Продолжительность жизни больных с третьим типом СМА составляет более 20 лет от начала дебюта заболевания.
Иногда выделяют четвертый тип СМА, при котором заболевание манифестирует в возрасте после 30 лет. Характеризуется постепенным и медленным нарастанием слабости и атрофическими изменениями мышц нижних конечностей, что в итоге приводит к инвалидности. Дыхательная мускулатура в патологический процесс обычно не вовлекается.
Это самый доброкачественный тип течения спинальной амиотрофии. Продолжительность жизни таких больных практически такая же, как и у здоровых людей.
Диагностика
Проявления проксимальной спинальной амиотрофии часто напоминают течение других неврологических и врожденных заболеваний, а также травматических повреждений структур спинного и головного мозга. Особенно затруднена диагностика этого заболевания у новорожденных и детей раннего возраста.
Ключевыми моментами в диагностике спинальной амиотрофии являются следующие исследования:
- Тщательный сбор анамнеза. Наличие случаев спинальной амиотрофии у родственников позволяет заподозрить это наследственное заболевание.
- Электронейромиография – специальное исследование нервно-мышечного аппарата. При этом исключается первичное поражение мышц и выявляются признаки, указывающие на патологию двигательных нейронов спинного мозга.
- Компьютерная и магнитно-резонансная томография. Эти методы позволяют иногда выявить атрофические изменения передних рогов спинного мозга. Однако, чаще они применяются для исключения другой патологии со стороны структур позвоночного столба и головного мозга.
- Биопсия мышц и с последующим гистологическим исследованием биоптата. Выявляются специфические изменения мышц, заключающиеся в чередовании пучковых атрофических и неизмененных мышечных волокон. Помимо этого, могут выявляться и компенсаторно гипертрофированные участки мышц, а также замещение мышечной ткани соединительной.
- Генетический анализ. Позволяет выявить точную причину заболевания: при исследовании ДНК выявляется мутация гена в пятой хромосоме.
Если в семье были случаи рождения детей со спинальной амиотрофией, при планировании последующей беременности супружеская пара направляется на консультацию к генетику. Также обязательной является дородовый анализ ДНК плода. Выявление синдрома Верднига-Гоффмана на стадии пренатальной диагностики служит показанием к прерыванию беременности.
Лечение
Учитывая наследственный генез, радикального лечения спинальной амиотрофии все еще не существует. Следовательно, эта патология считается неизлечимой.
На сегодняшний день таким больным проводится в основном симптоматическое лечение, направленное на поддержание функционирования мышечных волокон, а также – борьбу с осложнениями.
Основные принципы поддерживающей терапии при СМА:
- Специальное диетическое питание.
- Умеренные занятия лечебной физкультурой.
- Массаж.
- Некоторые физиотерапевтические методы (оксигенотерапия, ванны и т. д.).
- Ортопедические воздействия для предупреждения контрактур суставов.
- Медикаментозные препараты, улучшающие нервно-мышечную проводимость и питание мышц. Также назначаются средства, влияющие на метаболизм центральной нервной системы и микроциркуляцию.
При развитии дыхательной недостаточности, часто сопровождающей течение болезни (особенно, при первом и втором типе СМА), применяют различные методы вспомогательной или искусственной вентиляции легких. При выраженных нарушениях глотания может применяться гастростомическая питательная трубка. Многие больные с СМА вынуждены передвигаться с помощью инвалидных колясок.
В настоящее время ведутся научные исследования, направленные на создание препарата, увеличивающего синтез белка SMN, недостаточный уровень которого является причиной разрушения мотонейронов спинного мозга.
moyskelet.ru
Описание спинальной амиотрофии Верднига-Гоффмана
Спинальные мышечные атрофии, или СМА, подразумевают поражение нейронов спинного мозга, отвечающих за двигательные функции мускулов. Больше всего обычно страдает мускулатура ног и шеи, а вот мышцы верхних конечностей поражаются реже. У больных наблюдаются проблемы с передвижением, глотанием, удержанием головы, но при этом сохраняется чувствительность, и нет задержек в умственном развитии. Но если при остальных формах СМА у больных есть шанс дожить до старости, хоть и с инвалидностью, то при амиотрофии Верднига-Гоффмана максимальная продолжительность жизни не превышает 30 лет.
Поражение нейронов в спинном мозге приводит к тяжелым нарушениям в работе скелетных мышц
Патология встречается довольно редко – в одном случае из 80-100 тысяч. Но вот носителей гена, отвечающего за развитие аномалии, гораздо больше. Болезнь наследуется по рецессивно-аутосомному типу, и чтобы у ребенка проявилась амиотрофия Верднига-Гоффмана, оба родителя должны быть носителями гена. Хотя и в этом случае вероятность заболевания у малыша составляет только 25%. Причиной болезни является исключительно генетическая предрасположенность, никакой связи СМА с травмами, инфекциями и другими факторами не обнаружено.
Причины развития амиотрофии Верднига-Гоффмана
Проявления болезни
Специалисты выделяют три формы СМА Верднига-Гоффмана, отличающиеся между собой сроками проявления и типичными признаками.
Таблица. Формы СМА Верднига-Гоффмана
| Младенческая (СМА I типа) | С рождения до 6 мес. | От 6 мес. до 3 лет. |
| Ранняя детская (СМА II типа) | 7-10 мес. | 14-15 лет. |
| Поздняя (СМА III типа) | От 1,5 до 2,5 лет. | 20-30 лет. |
Все указанные формы патологии объединяет отсутствие каких-либо умственных и чувствительных нарушений, зато клиническая картина имеет значительные различия.
Последствия мутации гена СМН
Младенческая форма
При патологии I типа первые симптомы заметны уже при рождении ребенка: он появляется на свет с вялыми парезами, издает очень слабые крики, глубокие рефлексы отсутствуют. Для специалиста не составляет труда определить мышечную гипотонию, что позволяет с первых дней установить наличие атрофии Верднига-Гоффмана. Такие дети вяло сосут грудь, плохо глотают молоко, часто захлебываются. Даже движения языка затруднены, и при внимательном рассмотрении можно увидеть на нем непроизвольные сокращения мышц – мелкие волнообразные движения. Все это вызывает сложности с кормлением, ведь пища может проникнуть в дыхательные пути и вызвать гибель ребенка.
У грудничков с диагнозом СМА наблюдаются трудности с глотанием и дыхательными функциями
Кроме указанных симптомов наблюдается парез диафрагмы и деформации скелета: у ребенка может быть искривлен позвоночник, вдавлена или, наоборот, остро выступающая грудная клетка, вывернуты суставы. Дети с таким диагнозом сильно отстают в моторном развитии по сравнению со сверстниками. Они не могут удерживать голову, переворачиваться со стороны в сторону, тянуться за привлекающим внимание предметом и принимать сидячее положение. При этом мимику лица и функции глазных мышц болезнь не затрагивает, и эмоции, которые выражает ребенок, не искажаются.
К сведению: у ограниченного числа детей с амиотрофией Верднига-Гоффмана двигательные навыки все же проявляются, хоть и с большим опозданием, но затем через короткое время регрессируют.
СМА первого типа часто сопровождается другими врожденными патологиями:
- дисплазией суставов таза;
- гидроцефалией головного мозга;
- гемангиомами;
- пороком сердца.
Спинальная амиотрофия может развиваться одновременно с гидроцефалией
Развивается заболевание очень быстро, и летальный исход в большинстве случаев наступает в течение полугода после рождения. Некоторые малыши доживают до 2-3 лет. Причиной смерти, как правило, служит тяжелая форма сердечной и дыхательной недостаточности.
Ранняя форма
При втором типе амиотрофии дети в первые шесть месяцев своей жизни развиваются абсолютно нормально, без каких-либо настораживающих признаков. Некоторые в полгода даже начинают вполне активно подниматься на ноги и передвигаться вдоль кроватки или манежа. Первым симптомом является мышечная слабость, которая может развиваться постепенно или резко возникать на фоне любого детского заболевания, различных инфекций. Происходит это, как правило, на 7-10 месяце жизни малыша.
При амиотрофии второго типа первые 7-10 месяцев ребенок может развиваться абсолютно нормально
Функции мускулов сначала нарушаются в нижних конечностях, вследствие чего ребенок все хуже ползает, с трудом встает на ножки. Далее поражение распространяется все выше, что приводит к снижению глубоких рефлексов, появлению тремора пальцев, произвольных мускульных сокращений в языке, нарушению работы органов дыхания. Протекает данная форма патологии менее интенсивно, чем младенческая, и большинство больных доживает до подросткового возраста, хотя качество жизни все это время остается очень низким. Такие дети не могут себя обслуживать самостоятельно и без посторонней помощи им не обойтись. Причиной смерти обычно служат нарушения в работе дыхательных органов, а также различные инфекции, которые нередко поражают ослабленный болезнью организм.
Поздняя форма
Данную форму СМА специалисты считают наименее тяжелой, хотя и с ней человек может прожить максимум до 30 лет. Типичные проявления возникают чаще всего, когда ребенку исполнится 1,5-2 года. До этого времени физическое развитие происходит без малейших нарушений, малыш отлично ходит, бегает, проявляет нормальную для своего возраста активность. Лишь у ограниченного числа больных могут наблюдаться задержки с развитием моторики или излишняя медлительность.
У некоторых детей с поздней формой СМА может наблюдаться замедленное развитие мелкой моторики
Первые симптомы слабо выражены:
- ребенок быстрее устает;
- снижается координация движений, малыш чаще падает при беге и ходьбе;
- часто наблюдается вялость.
С прогрессированием болезни появляются другие признаки, более характерные:
- меняется походка, ребенок выше поднимает колени во время ходьбы;
- усиливается мышечная слабость;
- наблюдается мелкое дрожание пальцев рук;
- возникают произвольные спазмы языка, затруднения с глотательными функциями;
- развиваются деформации костей и суставов, особенно выражено это в грудной клетке.
С прогрессированием болезни усиливается деформация грудной клетки
Все эти процессы отличаются довольно медленным развитием, и способность ходить сохраняется примерно до 8-10 лет. В дальнейшем передвижение возможно лишь в инвалидной коляске, но способность к самообслуживанию полностью не утрачивается еще несколько лет. При наличии поддерживающей терапии люди с таким диагнозом доживают до 25-30 лет.
Диагностика заболевания
Синдром врожденной гипотонии мышц характерен не только СМА, но и ряду других патологий, например, ДЦП, амиотрофическому склерозу, миопатиям разных форм. Поэтому визуальный осмотр, а также информация о сроках появления симптомов и динамики их развития не помогут дифференцировать амиотрофию Верднига-Гоффмана. Исключить наличие других спинальных патологий позволяет проведение КТ или МРТ, но для определения СМА этих исследований недостаточно.
МРТ позволяет исключить наличие других спинальных патологий
Основным диагностическим методом при подозрении на спинальную атрофию мышц является электронейромиография, или ЭНМГ. Данный способ предназначен для анализа работы нервно-мышечной системы, в частности, прохождения нервных импульсов и реакции на них. Для подтверждения диагноза также назначается биопсия мышечных тканей и ДНК-исследования.
Важно! Людям, у которых в семьях уже наблюдались случаи заболевания СМА, рекомендуется пройти генетическое исследование на наличие SMN-гена, ответственного за нарушения развития мышц. Беременным женщинам проводят дородовый ДНК-анализ плода, и если диагноз подтвердится, это является серьезным показанием к прерыванию беременности.
Поддерживающая терапия
СМА Верднига-Гоффмана неизлечима, поэтому больным оказывается поддерживающая терапия, которая направлена на облегчение симптомов заболевания. Основной акцент делается на улучшение метаболизма в пораженных мышечных и нервных волокнах, что позволяет замедлить прогрессирование патологии. Для этого применяют медикаментозные средства нескольких групп: нейрометаболиты, препараты для облегчения нервно-мышечной передачи, препараты для улучшения трофики тканей и кровообращения.
Чаще всего назначаются:
- «Церебролизин»;
- «Ноотропил»;
Раствор для инъекций «Ноотропил»
- «Аминалон»;
- никотиновая кислота;
- «Скополамин»;
- «Нейромидин».
Таблетки «Нейромидин»
Вид препаратов, дозировки, длительность приема определяются лечащим врачом по результатам обследования и с учетом возраста ребенка. Помимо лекарств показана лечебная физкультура, физиотерапевтические процедуры, мягкий массаж. При усиленной деформации позвоночника применяются ортопедические корсеты и бандажи.
Как правило, спинальная амиотрофия не сопровождается нарушением координации и повышенной чувствительностью кожи, а развивается патология у всех пациентов примерно в одном и том же возрасте.
Разделяют 5 типов спинальной мышечной атрофии:
- Верднига-Гоффмана 1 — развивается до 1 года, зачастую приводит к летальному исходу.
- Верднига-Гоффмана 2 — развивается от 1 до 2-х лет, продолжительность жизни при этом не более 30 лет.
- Кульдберга-Веландер — это хроническая патология, развивающаяся с детства и до самой старости. Как правило, общая продолжительность жизни с этой болезнью более 40 лет.
- Кеннеди — развивается после 40-45 лет и до самой старости. Главное отличие — увеличивается грудь в объеме за счет набухания молочных желез.
- Шарко-Мари-Тута — это вялотекущее заболевание, проявляется в возрасте 15-30 лет и до самой старости. В большей степени болезнь поражает только мужское население. Отличается слабой прогрессией и поражает периферические нервы.
Причины развития
В 95% случаев данное заболевание передается по наследству. Проявление мышечной атрофии спины зависит от наличия рецессивного гена, который должен находиться и у отца, и у матери. Если ген передается только от одного родителя, то заболевание себя не проявляет.
Особые причины появления спинальной амиотрофии:
- ген заболевания находится в пятой хромосоме;
- не появляется самостоятельно, а только по генетической предрасположенности;
- спинальная амиотрофия встречается редко, но считается при этом распространенным наследственным заболеванием;
- если у обоих родителей присутствует данный ген, то вероятность появления амиотрофии у ребенка увеличивается до 25-50%;
- более 50% всех детей не доживают до 3-4 лет. Как правило, это связано с первой формой Верднигой-Гоффмана;
- впервые данное заболевание было обследовано в 1891 году Верднигом. Позже Гоффман в 1892 году составил подробное описания спинальной амиотрофии и пришел к выводу, что это абсолютно самостоятельное заболевание. На развитие мышечной атрофии не влияет питание, экология, стрессы или инфекции.
Симптоматика заболевания
Сейчас принято считать спинальную амиотрофию не единичной патологией, а группой заболеваний. Как правило, мышечная атрофия поражает поперечнополосатую мускулатуру шеи, головы и ног.
Заболевание это хоть и появляется на фоне генетической предрасположенности, но при этом обладает десятками симптомов:
- Истончение мышц и их слабость. Если заболевание появилось у детей, то в первую очередь поражаются мышцы ног, затем патология перекидывается на верхнюю часть туловища (на тазовый пояс, дыхательную систему, спину, шею, язык, глотку и голову). У взрослых заболевание проявляется на плечевом поясе и туловище, затем идет послабление ног и шеи.
- Произвольные движения нарушаются. Например, детям становится трудно держать голову, ползать или ходить. У взрослых присутствует быстрая утомляемость и затрудненное глотание.
- Нарушение координации и походки. Проявляется походка, которая схожа с механическими движениями как у заводной куклы. У человека можно наблюдать неестественное сгибание ног в коленях, создается ощущение пружинистой походки. Также проявляется утиная походка, когда человек переваливается с одной ноги на другую. Это связано со слабость мышц бедра и тазового пояса.
- Как правило, спинальная амиотрофия способна прогрессировать долгие годы. Сначала появляется слабость в мышцах, а хроническая форма сопровождается недееспособностью (невозможность вставать, ходить, держать голову, глотать).
- Из-за того, что поражаются нервные окончания, происходит фибрилляция мышц. Конечности и язык могут слегка, едва заметно, дергаться.
- Грудная клетка становится упругой и плотной. Это связано из-за расслабления глотательной и дыхательной мускулатуры.
- У детей развивается сколиоз и нарушение осанки. Ослабленные мышцы спины и поясницы не способны поддерживать позвоночный столб.
- Увеличиваются колени и голень. Гипертрофия проявляется на более поздних формах, когда тонкие мышцы кажутся большими и воспаленными. Иначе этот симптом называют псевдогипертрофией.
- В некоторых случаях увеличиваются в размере молочные железы, чаще всего у мужчин.
- Идет поражение рук, проявляется симметричное похудение и слабость, особенно у сгибателей.
Как диагностировать заболевание
При первых симптомах рекомендуется обратиться к педиатру или терапевту, который направит вас к невропатологу, неврологу и генетику. Они в свою очередь назначат необходимое обследование.
Этапы диагностирования:
- тщательный осмотр пациента. Сбор анамнеза. В этом пункте очень важно уточнить наследственную принадлежность. Врачи могут задавать вопросы о родителях и ближайших родственниках. Также стоит отметить примерный возраст появления мышечной слабости;
- невролог и невропатолог должны оценить состояние и тонус мышц. Внимательно изучают язык, процесс глотания. Для оценки состояния могут привлекать дополнительные процедуры, которые помогут увидеть с помощью прибора группы мышц, которые вовлечены в процесс силовых занятий;
- сдается анализ крови. Если происходит деструкция мышечных волокон, то отмечают повышение креатининфосфокиназы в крови;
- для определения зоны поражения нервных клеток проводится электронейромиография (ЭНМГ). Это обследование помогает выявить нарушение мышечных волокон и ее структуры;
- проводится рентгенография костей. При спинальной амиотрофии истончаются бедренные и плечевые кости, а также происходит искривление позвоночника;
- осмотр у генетика, а также проведение тестирования на изменения в таком гене, как 5q11 2-13 3.
Амиотрофия Верднига-Гоффмана
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. новорожденных. Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
Амиотрофия Верднига-Гоффмана имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю. Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию — амиотрофию Кугельберга-Веландера. Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Причины амиотрофии Верднига-Гоффмана
Амиотрофия Верднига-Гоффмана — наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы. Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) — ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена. Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах h5F5, NAIP, GTF2h3.
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы амиотрофии Верднига-Гоффмана
Врожденная форма (СМА I) клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов. Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают. Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов. У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия, косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры. Последний обуславливает развитие и прогрессирование дыхательной недостаточности, выступающей основной причиной летального исхода. В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии, которая может явиться смертельно опасным осложнением спинальной амиотрофии.
Ранняя детская форма (СМА II) дебютирует после 6-месячного возраста. К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять. Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов. Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана. Пациенты могут доживать до 15-летнего возраста.
Амиотрофия Кугельберга-Веландера (СМА III) – наиболее доброкачественная спинальная амиотрофия детского возраста. Манифестирует после 2-х лет, в отдельных случаях в период от 15 до 30 лет. Отсутствует задержка психического развития, длительное время пациенты способны самостоятельно двигаться. Некоторые из них доживают до глубокой старости, не теряя способности к самообслуживанию.
Диагностика амиотрофии Верднига-Гоффмана
В диагностическом плане для невролога имеет значение возраст появления первых симптомов и динамика их развития, данные неврологического статуса (в первую очередь наличие двигательных нарушений периферического типа на фоне абсолютно сохранной чувствительности), наличие сопутствующих врожденных аномалий и костных деформаций. Врожденная амиотрофия Верднига-Гоффмана может быть диагностирована неонатологом. Дифференциальный диагноз проводится с миопатиями, прогрессирующей мышечной дистрофией Дюшенна, боковым амиотрофическим склерозом, сирингомиелией, полиомиелитом, синдромом вялого ребенка, ДЦП, обменными заболеваниями.
С целью подтверждения диагноза проводится электронейромиография — исследование нервно-мышечного аппарата, благодаря которому выявляются характерные изменения, исключающие первично мышечный тип поражения и указывающие на патологию мотонейрона. Биохимический анализ крови не выявляет существенного повышения креатинфосфокиназы, характерного для прогрессирующей мышечной дистрофии. МРТ или КТ позвоночника в редких случаях визуализируют атрофические изменения передних рогов спинного мозга, но позволяют исключить другую спинальную патологию (гематомиелию, миелит, кисту и опухоль спинного мозга).
Окончательный диагноз «амиотрофия Верднига-Гоффмана» устанавливается после получения данных биопсии мышц и генетических исследований. Морфологическое изучение мышечного биоптата выявляет патогномоничную пучковую атрофию мышечных волокон с чередованием зон атрофии миофибрилл и неизмененной мышечной ткани, наличием отдельных гипертрофированных миофибрилл, участков соединительнотканных разрастаний. Проводимый генетиками ДНК-анализ включает прямую и косвенную диагностику. С помощью прямого метода возможна также диагностика гетерозиготного носительства генной аберрации, что имеет важное значение в генетическом консультировании сибсов (братьев и сестер) больных лиц, супружеских пар, планирующих беременность. При этом большую роль играет количественный анализ числа генов локуса СМА.
Дородовый анализ ДНК позволяет снизить вероятность рождения ребенка с болезнью Верднига-Гоффмана. Однако для получения ДНК-материала плода необходимо использовать инвазивные методы пренатальной диагностики: амниоцентез, биопсию хориона, кордоцентез. Амиотрофия Верднига-Гоффмана, диагностированная внутриутробно, выступает показанием к искусственному прерыванию беременности.
Причины возникновения спинальной мышечной атрофии
Содержание
- Причины возникновения спинальной мышечной атрофии
- Классификация патологии
- Болезнь Верднига-Гоффмана: симптоматика и прогноз
- Болезнь Кугельберга-Веландера: клиническая картина, выживаемость
- Диагностика, лечение и профилактика
Спинальная мышечная атрофия носит сугубо наследственный характер. Если у одного из родителей в организме присутствует пораженная патологическим изменением хромосома, то она неизменно передается по аутосомно-рецессивному типу и ребенку. Если оба родителя являются носителями патологически измененных хромосом, то амиотрофия у ребенка обнаруживается с самого младенчества. Как правило, у носителей таких хромосом заболевание никак не проявляется, а вот у их ребенка есть все признаки нарушения мышечной деятельности.
В поврежденных хромосомах отмечается мутация гена, который отвечает за синтез белка в организме. Такое нарушение становится причиной полной мышечной дисфункции. У больного со временем атрофируются мышцы, отвечающие за важные жизненные процессы. Постепенно пропадает дыхательный, глотательный рефлекс. Тонус мышц всего тела неизменно снижается, искажается лицо.
Когда амиотрофия появляется во взрослом возрасте, то это значит, что патологически измененная хромосома передалась от одного из родителей. Спинальная амиотрофия взрослых встречается только у мужчин, так как она скреплена с Х-хромосомой. Первые признаки данного заболевания у мужчин проявляются в зрелом и пожилом возрасте. Прожить с такой патологией взрослому можно достаточно долго. Однако необходимо постоянно поддерживать организм лекарственными препаратами, физиологическими процедурами и лечебной гимнастикой.
На развитие амиотрофии взрослых иногда влияют внешние факторы. Первые проявления болезни могут появиться как реакция на нарушенное кровообращение, несбалансированный рацион, проблемы с нервно-мышечной проводимостью, повреждения внутренних органов и их систем, курение, злоупотребление алкогольными напитками.
Исход данной патологии всегда летальный. Сколько проживет пациент, диагноз которому поставили во взрослом возрасте, зависит только от него самого. При правильном лечении и своевременной диагностике мотонейроны спинного мозга и ствола головного мозга отмирают не так быстро, как при отсутствии терапии.
Классификация патологии
Спинальные и невральные амиотрофии в научной медицине разделены на несколько типов. Они отличаются характером проявления, степенью выраженности симптомов, способностью больного к самостоятельным действиям. В зависимости от типа патологии врачи назначают соответственное лечение, скоординированное на замедление разрушительных реакций организма.
На сегодняшний день медики говорят о четырех основных типах спинальной мышечной атрофии, а именно:
- Болезнь Верднига-Гоффмана. Нарушение этого вида проявляется с первых дней жизни ребенка, поэтому его еще называют младенческой мышечной атрофией.
- Болезнь Дубовица. Это так называемый промежуточный вид, признаки которого обнаруживаются у ребенка от 7 месяцев до двух лет.
- Болезнь Кугельберга-Веландера. Этим термином называют ювенильный тип отклонения. Признаки впервые отмечаются у детей старшего возраста и неизменно прогрессируют.
- Взрослый тип болезни. От данного нарушения страдают мужчины старшего и пожилого возраста. Правильное лечение может замедлить разрушительные процессы и надолго продлить жизнь пациенту.
Все типы такого отклонения и нарушения функционирования нервной системы сходны в одном: вылечить их полностью невозможно. Быстрее всего болезнь забирает жизнь у малышей.
Если первые 2 типа считаются практически безнадежными, то болезнь Кугельберга-Веландера и амиотрофию во взрослом возрасте можно сдерживать благодаря лекарственным препаратам, специальным процедурам и лечебной физкультуре. Лечением подобных нарушений занимается только врач, народные средства бессильны.
При обнаружении первых признаков заболевания нужно обращаться к генетику, неврологу и невропатологу. Врачи проведут все необходимые обследования, поставят точный диагноз и расскажут пациенту или его родственникам о том, что нужно делать дальше. Соблюдая все рекомендации врачей, спинально-мышечную атрофию можно если не преодолеть, то существенно замедлить.
Существует также дистальная форма заболевания. Она встречается крайне редко. Основное ее отличие в том, что центр первичного поражения находится далеко от центра спинного мозга. Этот тип прогрессирует быстро, лечение дает слабый положительный результат.
Болезнь Верднига-Гоффмана: симптоматика и прогноз
Заболевание у детей встречается крайне редко. Как правило, оно диагностируется у одного ребенка из 100 тысяч детей до двух лет. Статистика гласит, что у 7 младенцев из 100 тысяч новорожденных проявляются первые симптомы уже с первого дня жизни вне утробы матери.
При диагностике обнаруживается, что клетки передних рогов спинного мозга недостаточно развиты. Черепные нервы нередко подвергаются патологическим изменениям. Скелетные мышцы еще сохраняют отдельные пучки здоровых нейронов, однако они подвергаются разрушению в течение короткого промежутка времени. У ребенка может отмечаться гиалиноз, гиперплазия соединительной ткани, нарушение целостности определенных мышечных волокон.
Медики выделяют три подвида данной болезни:
- врожденный;
- ранний детский;
- поздний детский.
Дети с врожденным подвидом СМА, как правило, не доживают до 9 лет. Уже с первых дней их жизни проявляются такие симптомы, как снижение тонуса мышц, полное отсутствие рефлексов. Со временем нарушается механизм сосания, дети тихо плачут, плохо глотают. Кроме того, больные не в состоянии самостоятельно пережевывать пищу.
По мере роста ребенка возникает парез диафрагмы, сколиоз, проблемы с суставами. Форма грудной клетки при этом сильно видоизменяется и деформируется. Кроме того, у больных детей нередко обнаруживаются признаки слабоумия и дефекты развития.
Врожденная форма очень быстро прогрессирует. К 8 годам больной ребенок превращается в полностью недееспособного человека. Когда дыхательный и глотательный рефлексы нарушаются окончательно, больной умирает от сердечной недостаточности, нехватки воздуха или проблем с пищеварением.
Ранняя детская форма болезни начинает развиваться со второго полугодия жизни. Летальный исход, как правило, происходит на 14 году жизни. Первые несколько месяцев младенец развивается нормально: держит головку, сидит, учится стоять. Однако потом проявляются те же признаки, что и при врожденной форме. Данный тип развивается мягче и не так агрессивен, как врожденный тип. Однако летальный исход наступает в любом случае.
Признаки поздней формы начинают проявляться к 2 годам. Заболевание развивается постепенно и мягко. Сначала ребенок может даже ходить и бегать, но далее эти навыки исчезают. Люди с такой формой могут прожить в среднем до 30 лет.
Болезнь Кугельберга-Веландера: клиническая картина, выживаемость
Спинальная амиотрофия Кугельберга-Веландера отличается от болезни Верднига-Гоффмана тем, что является относительно доброкачественным процессом. Это значит, что развивается данная патология очень медленно и дает человеку возможность прожить практически до самой старости. При этом больной долгое время сохраняет относительную дееспособность. Человек с данным расстройством может самостоятельно передвигаться, ходить на работу, за покупками и т.д.
У больных есть возможность до определенного возраста, пока заболевание не распространилось по всему телу, вынашивать и рожать детей. Однако есть большая вероятность, что нарушение передастся по наследству. Если один из партнеров здоров, это также не является гарантией того, что дети будут здоровы. Планирование беременности в таких случаях должно сопровождаться консультацией у генетика, так как патологически измененные хромосомы можно увидеть у ребенка уже на ранних сроках беременности.
Ученый-медик Веландер указывал на то, что первые признаки болезни проявляются после двухлетнего возраста. Пик происходит, как правило, между вторым и пятым годом жизни. Однако у некоторых людей болезнь обнаруживает себя гораздо позже. Это случается и в юношеском возрасте, даже если до этого никаких проблем с опорно-двигательным аппаратом не наблюдалось.
Первыми тревожными симптомами являются ситуации, когда ребенок часто спотыкается, ему сложно ходить по лестнице, при ходьбе у него подворачиваются или подгибаются коленки. Позже может обнаружиться сколиоз, деформация грудной клетки, тремор рук, судороги нижних конечностей.
Поначалу болезнь задевает только нижние конечности. В более позднем возрасте нарушается моторика верхней части туловища. Однако человек сохраняет подвижность практически до самой старости. Залогом долгой жизни в этом случае является специальная лечебная гимнастика, отказ от сидячего образа жизни и всех вредных привычек, сбалансированное питание, полноценный сон, ежедневные прогулки на свежем воздухе.
Важно помнить, что атрофия Кугельберга не является причиной для полной инвалидности. Люди с таким нарушением неполноценны, однако они могут вести привычный образ жизни и обходиться без посторонней помощи на протяжении долгих лет со дня появления первых симптомов.
- https://spina-expert.ru/drugie-bolezni/spinalnaya-amiotrofiya-verdniga-goffmana/
- http://SpineDoc.ru/patologii-myshc/spinalnaya-amiotrofiya.html
- http://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/Werdnig-Hoffmann
- https://spinazdorov.ru/muscle/spinalnaya-myshechnaya-atrofiya.html
zdorovo.live
Что такое спинальная амиотрофия Верднига Гоффмана
Спинальная амиотрофия Верднига Гоффмана передается по наследству и это злокачественное заболевание нервной системы. Происходит слабость практически всех мышечных волокон. Пациент не может самостоятельно сидеть и передвигаться. На сегодняшний день не существует эффективных способ лечения патологии.
Как правило, заболевание выявляется с рождения и до полутора лет. Это самая тяжелая форма мышечной атрофии при которой развиваются парезы. Заболевание встречается очень редко, примерно один случай на десять тысяч человек.
Симптомы
Существует четыре вида спинальной амиотрофии, они отличаются симптомами и длительностью жизни больного. Все формы патологии проявляются общим признаком, это нарушение умственных функций и чувствительности.
В области тазовых органов не происходят изменения. Все признаки проявляются с нарушением двигательной системы.
Спинальная амиотрафия 1 типа
Происходит нарушение глотательной и сосательной функции. Ребенку становится трудно двигать языком и заметно, как возникает волнообразное сокращение на нем. Крики малыша слышно слабо. Если снижаются глотательные функции, то будут проблемы с питанием, так как еда будет попадать в дыхательную систему. Как правило, это приводит к тому, что развивается аспирационная пневмония и малыш из-за этого может умереть.
Если происходит поражение межреберных мышц, то будут нарушения дыхательной системы. По началу это может быть не так заметно, но со временем состояние ребенка будет ухудшаться. Как правило, мышца лица, которые отвечает на движение глаз не нарушаются. Ребенок не сидит, не может держать и поворачивать голову и тянуться за игрушками. Если были какие-то движения развиты до спинальной амиотрофии, то они пропадут.
Кроме этих нарушений возникает и деформация грудного отдела. Если заболевание заметно стало сразу же после того, как родился ребенок, то он живет не больше полугода. Если же развитие патологии было после трех месяцев, то малыш проживет примерно два года. Летальный исход может быть куда раньше, даже из-за инфекционного поражения дыхательной системы. Спинальная амиотрофия Верднига может быть вместе с другими врожденными патологиями.
Спинальная амиотрафия 2 типа
Заболевание у малыша развивается от полугода и до двух лет. До этого момента не заметно никаких изменений. Ребенок самостоятельно держит голову, сидит, переворачивается и даже ходит.
Дальше происходит небольшая слабость в мышцах, как правило, возникает это с бедер. Понемногу ребенок начинает трудно ходить и у него снижается реакция сухожилий. Слабость в мышцах развивается медленно, но со временем происходит мышечная атрофия. Также нарушается мускулатура дыхательной системы.
Как при первом типе спинальной амиотрофии не происходит поражение мышц глаз и мимических. Может наблюдаться дрожь в кистях, подёргивание языка, рук и ног. Дальше развивается мышечная слабость в шее и это приводит к тому, что голова свисает. Может также быть деформация грудного отдела, сколиоз. Форма заболевания является доброкачественной и чаще всего могут быть нарушение дыхательной системы в подростковом возрасте.
Спинальная амиотрофия 3 типа
Часто болезнь встречается у больных в возрасте от двух и до пятнадцати лет. Симптомы проявляются в виде неправильной походки и мышечной слабости в конечностях.
Дальше происходит снижение тонуса в ногах и на этом фоне развивается атрофия. Такие изменения могут быть незаметны, так как в таком возрасте хороший подкожный жировой слой. Ребенок начинает постоянно спотыкаться, падать. Со временем больному тяжело передвигаться и в итоге он перестает ходить.
Позже может происходить и поражение верхних конечностей. Тогда возникает нарушение мимических мышц, а глазами пациент двигает без проблем. Пропадает рефлекс тех мышц, которые уже поражены. Может возникать деформация скелета и суставов. При таком заболевании, если будет необходимое лечение больной доживает примерно до сорока лет.
Спинальная амиотрофия 4 типа
Данный вид спинальной амиотрофии происходит уже во взрослом возрасте после 35 лет. Проявляется патология в виде мышечной слабости в конечностях и снижения рефлексов.
Дальше развивается на этом фоне атрофия мышц, это приводит к утрате подвижности ног. Дыхательная система в этом случае не страдает. Больной может жить с таким заболеванием, как обычный здоровый человек. Такой вид патологии самый доброкачественный по сравнению с другими.
Диагностика
На раннем развитии заболевание бывает тяжело поставить точный диагноз, так как признаки похожи с другими болезнями. Первым делом ребенок должен пройти консультацию у невропатолога. Если у малыша заболевание при рождении, то примерный диагноз могут поставить в роддоме. Специалист проводит осмотр и проверяет нарушения двигательной системы.
Проводятся следующие исследования:
- Магнитно-резонансная томография назначается для проверки позвоночника. Она обязательно используется при спинальной амиотрофии, потому как позволяет понять, в каком состоянии находится интересующая область. Процедура считается безопасной, потому как она не ухудшает состояние здоровья человека. Более того, она рекомендована даже для детей, у которых имеется амиотрофия Верднига. Если специалисты и родители понимают, что несовершеннолетний не сможет неподвижно лежать около часа, тогда может возникнуть вопрос о применении наркоза. В любом случае, от МРТ не стоит отказываться, она позволит узнать немало полезной информации о состоянии здоровья и о развитии амиотрофии Верднига.
- Электронейромиогафия помогает изучить состояние нервных и мышечных окончаний. Это тоже требуется для того, чтобы понять, насколько тяжело проходит заболевание. Если у человека спинальная амиотрофия, тогда важно как можно больше собрать информации о состоянии организма. В частности придётся проходить данное обследование.
- Генетическая диагностика дает возможность выявить мутацию гена. Это актуально для тех случаев, когда имеется амиотрофия Верднига. Конечно, обследование не самое простое, и не во всех клиниках его могут провести. При этом оно является обязательным для тех людей, которые столкнулись со спинальной амиотрофией.
Найти врожденную патологию можно еще до рождения малыша. Проводится диагностика в том случае, если у девушки наблюдается слабое шевеление плода. Тогда беременная должна лечь в больницу для полного обследования.
Диагностика ДНК проводится не только для выявления патологии у малыша, но и вовремя вынашивания до 38 недель. Если есть подозрения на врожденные патологии у ребенка, то лучше всего пройти обследование еще до родов.
Лечение
Спинальная амиотрофия Верднига — это генетическое заболевание, которое полностью не лечится. Терапия дает возможность поддержать жизненные функции, чтобы не допустить осложнений. По этой причине не стоит закрывать на то, в каком состоянии находится человек. Обязательно придётся принять меры для того, чтобы поддерживать самочувствие в максимально хорошем виде.
Как правило, специалист назначает лекарственные препараты, массажные процедуры, физиотерапию. Только в комплексе удаётся справиться со спинальной амиотрофией. Исходить придётся от индивидуальных особенностей организма больного.
Потому как важно правильно подобрать те меры, которые могут ослабить клинические проявления амиотрофии Верднига. Если соблюдать все рекомендации врача, то это поможет не ухудшить состояние больного. Более того, при правильном подходе здоровье ещё и улучшится.
Назначаются следующие препараты при спинальной амиотрофии:
- Медикаменты Нивалин, Прозерин помогают улучшить проходимость импульсов. Это обязательно требуется для тех случаев, когда у человека возникает спинальная амиотрофия.
- Средства Оротат калия, Лидаза, Никотиновая кислота нормализуют обменный процесс и улучшают кровоток. При этом крайне важно активировать кровообращение, потому как от него зависит состояние всего организма. Дозировка препаратов назначается в зависимости от самочувствия человека. Также может потребоваться подобрать конкретные средства исходя из того, насколько хорошо конкретные препараты помогают человеку.
- Витамины группы В Мильгамма, Нейромультивит улучшают иннервацию мышц. Также они в целом укрепляют организм, а это необходимо при спинальной амиотрофии. Можно однозначно сказать, что витаминные комплексы не будут лишними. При этом их плюс в том, что они не вредят здоровью.
- Ноотропные средства Ноотропил, Пиратецам помогают привести в норму кровоснабжение. Также они способствуют улучшению функции ЦНС и активируют мозговую деятельность. Когда диагностирована амиотрфия Верднига, тогда без препаратов данной группы не обойтись.
Употреблять лекарственные средства необходимо с осторожностью и под наблюдением врача, так как имеется ряд побочных действий. Как известно, маленькая вероятность выживаемости среди людей, которые болеют амиотрофией Верднига. Новорожденные с врожденной патологией имеют небольшое участие со стороны врача. Если происходит сильное ослабление мышц, то могут делать шинирование.
Если при спинальной амиотрофии происходит сколиоз, то это приводит к искривлению позвонков и считается тяжелой формой болезни. Операция должна быть только в том случае, если врачи увидят в этом необходимость. Обязательно нужно соблюдать диету, так как это играет важную роль в лечении амиотрофии Верднига.
Если вовремя начать лечение, тогда амиотрофия Верднига будет хотя бы немного легче переноситься человеком. Именно поэтому люди должны незамедлительно обратиться за врачебной помощью и побеспокоиться о том, чтобы пройти полную диагностику. Как уже говорилось, амиотрофия Верднига полностью не лечится. При этом потребуется бороться с её проявлениями, чтобы можно было улучшить состояние пациента, который страдает от болезни.nevrology.net
24.1.2. Нейрогенные амиотрофии
Спинальная амиотрофия Верднига–Гоффманна. Заболевание описано Дж. Верднигом в 1891 г. и Ж. Гоффманном в 1893 г. Частота 1 на 100 000 населения, 7 на 100 000 новорожденных. Наследуется по аутосомно‑рецессивному типу.
Патоморфология. Обнаруживаются недоразвитие клеток передних рогов спинного мозга, демиелинизация передних корешков. Часто имеются аналогичные изменения в двигательных ядрах и корешках V, VI, VII, IX, X, XI и XII черепных нервов. В скелетных мышцах нейрогенные изменения характеризуются «пучковой атрофией», чередованием атрофированных и сохранных пучков мышечных волокон, а также нарушениями, типичными для первичных миопатий (гиалиноз, гипертрофия отдельных мышечных волокон, гиперплазия соединительной ткани).
Клинические проявления. Выделяют три формы заболевания: врожденную, раннюю детскую и позднюю, различающиеся временем проявления первых клинических симптомов и темпом течения амиотрофического процесса.
При врожденной форме с первых дней жизни у детей выражены генерализованная мышечная гипотония и гипотрофия мышц, снижение либо отсутствие сухожильных рефлексов. Рано определяются бульбарные расстройства, проявляющиеся вялым сосанием, слабым криком, фибрилляциями языка, снижением глоточного рефлекса. Заболевание сочетается с костно‑суставными деформациями: сколиозом, воронкообразной или «куриной» грудной клеткой, контрактурами суставов. Развитие статических и локомоторных функций резко замедлено. Лишь у ограниченного числа детей с большим опозданием формируется способность держать голову и самостоятельно садиться. Однако приобретенные двигательные навыки быстро регрессируют. У многих детей с врожденной формой болезни снижен интеллект. Часто наблюдаются врожденные пороки развития: врожденная гидроцефалия, крипторхизм, гемангиома, дисплазия тазобедренных суставов, косолапость и др.
Течение. Болезнь имеет быстро прогрессирующее течение. Летальный исход наступает до 9‑летнего возраста. Одной из основных причин смерти являются тяжелые соматические расстройства (сердечно‑сосудистая и дыхательная недостаточность), обусловленные слабостью мускулатуры грудной клетки и снижением участия ее в физиологии дыхания.
При ранней детской форме первые признаки болезни возникают, как правило, на втором полугодии жизни. Моторное развитие в течение первых месяцев удовлетворительное. Дети своевременно начинают держать голову, сидеть, иногда стоять. Заболевание развивается подостро, нередко после инфекции, пищевой интоксикации. Вялые парезы первоначально локализуются в ногах, затем быстро распространяются на мышцы туловища и руки. Диффузные мышечные атрофии сочетаются с фасцикуляциями, фибрилляциями языка, мелким тремором пальцев, сухожильными контрактурами. Мышечный тонус, сухожильные и надкостничные рефлексы снижаются. В поздних стадиях возникают генерализованная мышечная гипотония, явления бульбарного паралича.
Течение. Злокачественное, хотя и мягче по сравнению с врожденной формой. Летальный исход наступает к 14–15 годам жизни.
При поздней форме первые признаки болезни возникают в 1,5– 2,5 года. К этому возрасту у детей полностью завершено формирование статических и локомоторных функций. Большинство детей самостоятельно ходят и бегают. Заболевание начинается незаметно. Движения становятся неловкими, неуверенными. Дети часто спотыкаются, падают. Изменяется походка: они ходят, сгибая ноги в коленях (походка «заводной куклы»). Вялые парезы первоначально локализуются в проксимальных группах мышц нижних конечностей, в дальнейшем сравнительно медленно переходят на проксимальные группы мышц верхних конечностей, мышцы туловища; атрофии мышц обычно малозаметны вследствие хорошо развитого подкожного жирового слоя. Типичны фасцикуляции, мелкий тремор пальцев, бульбарные симптомы – фибрилляции и атрофия языка, снижение глоточного и небного рефлексов. Сухожильные и надкостничные рефлексы угасают уже в ранних стадиях болезни. Костно‑суставные деформации развиваются параллельно основному заболеванию. Наиболее выражена деформация грудной клетки.
Течение. Злокачественное, но мягче, чем у первых двух форм. Нарушение способности самостоятельной ходьбы происходит в 10–12‑летнем возрасте. Больные живут до 20–30 лет.
Диагностика и дифференциальный диагноз. Диагноз строится на основании данных генеалогического анализа (аутосомно‑рецессивный тип наследования), особенностей клиники (раннее начало, наличие диффузных атрофии с преимущественной локализацией в проксимальных группах мышц, генерализованной мышечной гипотонии, фасцикуляций и фибрилляций языка, отсутствие псевдогипертрофий, прогредиентное и в большинстве случаев злокачественное течение и др.), результатах глобальной (накожной) и игольчатой электромиографии и морфологического исследования скелетных мышц, позволяющего выявить денервационный характер изменений.
Дифференцировать врожденную и раннюю формы следует в первую очередь от заболеваний, входящих в группу синдромов с врожденной мышечной гипотонией (синдром «вялого ребенка»): амиатонии Оппенгейма, врожденной доброкачественной формы мышечной дистрофии, атонической формы детского церебрального паралича, наследственных болезней обмена веществ, хромосомных синдромов и др. Позднюю форму следует дифференцировать от спинальной амиотрофии Кугельберга–Веландера, прогрессирующих мышечных дистрофий Дюшенна, Эрба–Рота и др.
Лечение. При спинальной амиотрофии Верднига–Гофмана назначают ЛФК, массаж, препараты, улучшающие трофику нервной ткани – церебролизин, аминалон (гаммалон), пиридитол (энцефабол).
Спинальная юношеская псевдомиопатическая мышечная атрофия Кугельберга–Веландера. Частота не установлена. Наследуется по аутосомно‑рецессивному, реже – по аутосомно‑доминантному, рецессивному сцепленному с Х‑хромосомой типу.
Патоморфология. Обнаруживаются недоразвитие и дегенерация клеток передних рогов спинного мозга, демиелинизация передних корешков, дегенерация двигательных ядер IX, X, XII черепных нервов. В скелетных мышцах – сочетанные изменения, типичные для нейрогенных амиотрофии (пучковая атрофия мышечных волокон) и первичных миодистрофий (атрофии и гипертрофии мышечных волокон, гиперплазия соединительной ткани).
Клинические проявления. Первые признаки заболевания проявляются в 4–8 лет. Описаны случаи начала болезни и в более позднем возрасте – 15–30 лет. В начале болезни характерными симптомами являются патологическая мышечная утомляемость в ногах при длительной физической нагрузке (ходьба, бег), иногда спонтанные подергивания мышц.
Внешне обращают на себя внимание увеличенные икроножные мышцы. Атрофии первоначально локализуются в проксимальных группах мышц нижних конечностей, тазового пояса, бедер и всегда симметричны. Их появление вызывает ограничение двигательных функций в ногах – затруднение при подъеме на лестницу, вставании с горизонтальной поверхности. Постепенно изменяется походка. В стадии выраженных двигательных расстройств она приобретает характер «утиной». Атрофии в проксимальных группах мышц верхних конечностей обычно развиваются спустя несколько лет после поражения нижних конечностей. Вследствие атрофии лопаточной и плечевой областей уменьшается объем активных движений в руках, лопатки становятся «крыловидными». Мышечный тонус в проксимальных группах мышц снижается. Сухожильные рефлексы угасают вначале на ногах, а затем на руках (рефлексы с двуглавой и трехглавой мышц плеча). Характерными симптомами, отличающими спинальную амиотрофию Кугельберга–Веландера от фенотипически сходной первичной прогрессирующей мышечной дистрофии Эрба–Рота, являются фисцикуляции мышц, фибрилляций языка, мелкий тремор пальцев. Костно‑суставные нарушения, сухожильные ретракции выражены умеренно либо отсутствуют.
Течение. Болезнь медленно прогрессирует.
Диагностика и дифференциальный диагноз. Диагноз ставится на основании данных генеалогического анализа (аутосомно‑рецессивный, аутосомно‑доминантный, рецессивный сцепленный с Х‑хромосомой тип наследования), особенностей клиники (начало болезни преимущественно в возрасте 4–8 лет, симметричные атрофии мышц, распространяющиеся по восходящему типу фасцикуляции мышц, мелкий тремор языка, псевдогипертрофии икроножных мышц, медленое прогредиентное течение), результатов глобальной и игольчатой электромиографии и морфологического исследования скелетных мышц, позволяющего выявить денервационный характер изменений.
Дифференцировать болезнь следует от прогрессирующих мышечных дистрофий Беккера, Эрба–Рота, спинальной амиотрофии Верднига–Гоффманна.
Наследственная дистальная спинальиая амиотрофия. Частота не установлена. Наследуется по аутосомно‑рецессивному, реже – по аутосомно‑доминантному, рецессивному сцепленному с Х‑хромосомой типу.
Патоморфология. Соответствует другим спинальным амиотрофия м.
Клинические проявления. Первые признаки заболевания проявляются преимущественно в первой декаде жизни. Начальными симптомами болезни являются слабость и атрофия дистальной мускулатуры ног. В 25 % случаев наблюдаются слабость и атрофия дистальной мускулатуры рук. Отличительные особенности – грубые деформации стоп, ранняя утрата ахиллова рефлекса при сохранности коленных и глубоких рефлексов с рук, отсутствие чувствительных расстройств.
Течение. Болезнь медленно прогрессирует.
Диагностика и дифференциальный диагноз. Диагноз ставится на основании генеалогического анализа (аутосомно‑доми‑нантный, аутосомно‑рецессивный, рецессивный сцепленный с Х‑хромосомой тип наследования), особенностей клинической картины (начало в первой декаде жизни, преимущественная локализация атрофии в дистальных отделах нижних конечностей, грубые деформации стоп, отсутствие чувствительных нарушений, медленное прогрессирование миодистрофического процесса), результатов глобальной и игольчатой электромиографии, позволяющей выявить вовлечение в процесс передних рогов спинного мозга.
Дифференцировать заболевание следует от дистальной миопатии Говерса–Веландера, невральной амиотрофии Шарко–Мари–Тута.
Невральная амиотрофия Шарко–Мари–Тута. Частота 1 на 50 000 населения. Наследуется по аутосомно‑доминантному, реже – по аутосомно‑рецессивному сцепленному с Х‑хромосомой типу.
Патоморфология. Обнаруживается сегментарная демиелинизация в нервах, в мышцах – денервация с явлениями «пучковой» атрофии мышечных волокон.
Клинические проявления. Первые признаки заболевания чаще проявляются в 15–30 лет, реже в дошкольном возрасте. В начале болезни характерными симптомами являются мышечная слабость, патологическая утомляемость в дистальных отделах нижних конечностей. Больные быстро устают при длительном стоянии на одном месте и нередко для уменьшения утомления в мышцах прибегают к ходьбе на месте («симптом топтания»). Реже заболевание начинается с чувствительных расстройств – болей, парестезии, ощущения ползания мурашек. Атрофии первоначально развиваются в мышцах голеней и стоп. Мышечные атрофии, как правило, симметричны. Поражаются перонеальная группа мышц и передняя большеберцовая мышца. Вследствие атрофии ноги резко сужаются в дистальных отделах и приобретают форму «перевернутых бутылок» или «ног аиста». Стопы деформируются, становятся «выеденными», с высоким сводом. Парез стол изменяет походку больных. Они ходят, высоко поднимая ноги: ходьба на пятках невозможна. Атрофии в дистальных отделах рук – мышцах тенара, гипотенара, а также в мелких мышцах кистей присоединяются спустя несколько лет после развития амиотрофических изменений в ногах. Атрофии в кистях симметричны. В тяжелых случаях при выраженных атрофкях кисти приобретают форму «когтистых», «обезьяньих». Мышечный тонус равномерно снижен в дистальных отделах конечностей. Сухожильные рефлексы изменяются неравномерно: ахилловы рефлексы снижаются в ранних стадиях болезни, а коленный рефлекс, рефлексы с трех– и двуглавой мышц плеча длительное время остаются сохранными. Чувствительные расстройства определяются нарушениями поверхностной чувствительности по периферическому типу («тип перчаток и носков»). Часто имеются вегетативно‑трофические нарушения – гипергидроз и гиперемия кистей и стоп. Интеллект обычно сохранен.
Течение. Болезнь медленно прогрессирует. Прогноз в большинстве случаев благоприятен.
Диагностика и дифференциальный диагноз. Диагноз строится на основании данных генеалогического анализа (аутосомно‑доминантный, аутосомно‑рецессивный, рецессивный сцепленный с Х‑хромосомой тип наследования), особенностей клиники (атрофии дистальных отделов конечностей, расстройства чувствительности по полиневритическому типу, медленное прогрессирующее течение), результатов глобальной, игольчатой и стимуляционной электромиографии (снижение скоростей проведения по чувствительным и двигательным волокнам периферических нервов) и в ряде случаев биопсии нервов.
Дифференцировать заболевание следует от дистальной миопатии Говерса–Веландера, наследственной дистальной спинальной амиотрофии, миотонической дистрофии, периферических нейропатий, интоксикационных, инфекционных полиневритов и других болезней.
Лечение. Терапия прогрессирующих нервно‑мышечных заболеваний направлена на улучшение трофики мышц, а также проводимости импульсов по нервным волокнам.
С целью улучшения трофики мыши назначают аденозинтрифосфорную кислоту, кокарбоксилазу, церебролизин, рибоксин, фосфаден, карнитина хлорид, метнонин, лейцин, глутаминовую кислоту. Анаболические гормоны назначают только в виде коротких курсов. Применяют витамины Е, А, группы В и С. Показаны средства, улучшающие микроциркуляцию: никотиновая кислота, ксантинол никотинат, никошпан, пентоксифиллин, пармидин. Для улучшения проводимости назначают антихолинэстеразные препараты: галантамин, оксазил, пиридостигмина бромид, стефаглабрина сульфат, амиридин.
Наряду с медикаментозной терапией применяют лечебную физкультуру. массаж и физиотерапию. Важна профилактика костно‑суставных деформаций и контрактур конечностей.
В комплексном лечении больных используют следующие виды физиотерапии: электрофорез лекарственных средств (прозерин, хлорид кальция), диадинамические токи, миостимуляцию синусоидальными модулированными токами, электростимуляцию нервов, ультразвук, озокерит, грязевые аппликации, радоновые, хвойные, сульфидные и сероводородные ванны, оксигенобаротерапию. Показано ортопедическое лечение при контрактурах конечностей, умеренной деформации позвоночника и асимметричном укорочении конечностей. Показаны полноценные белки, калиевая диета, витамины.
Лечение должно быть индивидуальным, комплексным и продолжительным, состоять из последовательных курсов, включающих сочетание различных видов терапии.
studfiles.net