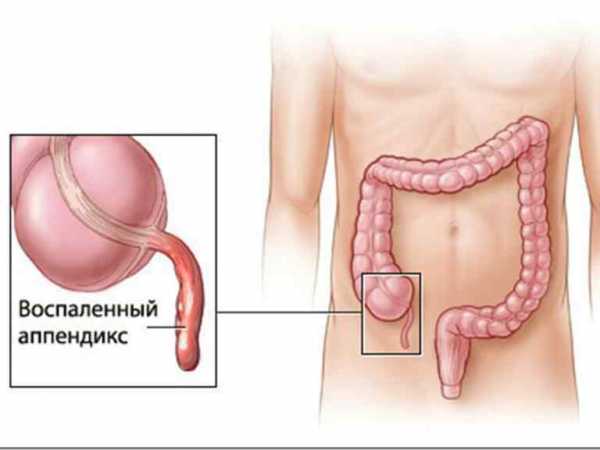
Лечение суставов - артроз, артрит, остеохондроз и многое другое
Затылочная эпилепсия у детей
Затылочная эпилепсия у детей
А Б В Г Д Е Ж З И К Л М Н О П Р С Т У Ф Х Ц Ч Ш Щ Э Ю ЯЗатылочная эпилепсия принадлежит к гетерогенной группе заболеваний, начальные клинические и электрофизиологические проявления которых свидетельствуют о фокальном характере эпилептических припадков. Локализация эпилептогеннных очагов размещается в затылочной доле вследствие первичного поражения. Заболевание может дебютировать в любом возрасте.
Затылочная эпилепсия может развиваться при конституцианальнойэпипредиспозиции или возникнуть, как наследственное заболевание.
Причинами симптоматических затылочных эпилепсий являются дисгенезии мозга (фокальная корковая дисплазия, микрогирия и др.), опухоли, нейрофиброматоз, киста затылочной доли, врожденные пороки развития мозга, синдром MELAS (митохондриальная энцефаломиопатия, лактатацидоз, разорванные красные волокна), нейроинфекции.
При затылочной эпилепсии происходят изменения нейрональной активности головного мозга. При этом наблюдается непредвиденная выраженная деполяризация нейронов в мозге, которая ведет к существенным нарушениям процессов таламокортикального взаимодействия и повышению чувствительности кортикальных нейронов. Напоявление приступов влияет излишнее выделение возбуждающих нейротрансмиттеров – аспартата и глутамата, а также нехватка тормозных нейромедиаторов, прежде всего, ГАМК.
Затылочная эпилепсия, как и теменная, характеризуется преимущественно простыми парциальными пароксизмами без нарушения сознания. Клинические ее проявления подразделяются на начальные и последующие симптомы. Начальные клинические симптомы обусловлены эпилептической активностью непосредственно в затылочной доле, тогда как последующие симптомы являются результатом распространения эпилептической активности на другие области мозга. К начальным клиническим симптомам затылочных пароксизмов относятся простые зрительные галлюцинации, пароксизмальный амавроз, пароксизмальные нарушения полей зрения, субъективные ощущения в области глазных яблок, моргание, девиация головы и глаз. Простые зрительные галлюцинации представляют собой яркие вспышки света перед глазами, светящиеся пятна, круги, звезды, квадраты, прямые или зигзагообразные линии. Они могут быть как одного цвета, так и многоцветными, неподвижными или перемещающимися в поле зрения горизонтально, вращательно или приближаться и удаляться. Простые зрительные галлюцинации, как изолированный симптом или как часть более сложного приступа, всегда указывают на локализацию эпилептогенного очага в затылочной доле.
Пароксизмальныйамавроз проявляется в виде нечеткости или временной утраты зрения. Больные обычно описывают свои ощущения как «черноту перед глазами», реже – как «белую пелену перед глазами». Пароксизмальный амавроз нередко сочетается с простыми зрительными галлюцинациями, а иногда – с мигренозной головной болью. Пароксизмальные нарушения полей зрения встречаются относительно редко в виде пароксизмальной гемианопсии или квадрантной гемианопсии в течение нескольких секунд или нескольких минут. Субъективные ощущения в области глазных яблок выражаются преимущественно чувством движения глаз при отсутствии объективных симптомов. Данный признак является типичным для затылочной эпилепсии. Моргание при эпилепсиях отмечается в самом начале приступа, имеет насильственный характер и напоминает трепетание крыльев бабочки. Девиация головы и глаз характерна для эпилептических пароксизмов различной локализации. При затылочной эпилепсии девиация головы и глаз нередко является одним из начальных симптомов приступа, при этом голова и глаза поворачиваются в противоположную сторону. Эпилептическая активность часто распространяется на другие области мозга, вследствие чего возникают различные клинические симптомы.
В диагностировании используется межприступная ЭЭГ (электроэнцефалограмма) с применением поверхностных электродов, данное исследование является достаточно информативным. Однако в ряде случаев регистрируются фокальные эпилептические образцы, преимущественно в затылочной или задневисочной области. Наличие эпилептических образцов в задневисочных областях может свидетельствовать о возможной затылочной природе эпилептических пароксизмов.
MPT-исследование проводится для исключения локальной патологии головного мозга. МРТ обязательно при впервые возникших приступах затылочной эпилепсии, имеет высокую диагностическую значимость и позволяет в большинстве случаев идентифицировать патологические изменения в затылочной доле.
Нейровизуализация направлена на выявление неэпилептогенных органических дефектов типа вентрикуломегалии, арахноидальных кист.
Нейропсихологическое исследование проводиться с целью определения речевых и поведенческих расстройств, зрительных отклонений, снижения внимания и нарушений памяти.
Диагноз ставят после проведения выше упомянутых исследований и дифференциальной диагностики, которая проводится при ранней форме на основание нарушений кровообращения мозга.
Медикаментозное лечение при затылочной эпилепсии показано при наблюдении частых приступов или нарушении качества жизни больного. Приступы часто влияют на психосоциальное поведение ребенка.
Препараты выбора при парциальных припадках (без вторичной генерализации или вторично-генерализованных) – карбамазепины и валъпроаты. В целом при парциальных припадках ряд препаратов (карбамазепины, вальпроаты, фенитоин, фенобарбитал) достаточно эффективны, однако фенобарбитал и фенитоин не являются препаратами выбора вследствие побочного действия.При резистентности к карбамазепинам и вальпроатам или плохой их переносимости применяют новые противоэпилептические препараты, наиболее эффективный из них при парциальных формах эпилепсии топирамат (топамакс). Топамакс назначается детям старше 2 лет при монотерапии в первую неделю лечения в дозе 0,5-1 мг/кг в сутки (суточную дозу делят на 2 приема). Детям старше 2 лет при монотерапии топамаксом рекомендуются дозы 3-6 мг/кг в сутки. Детям старшего возраста и подросткам в начале монотерапии топамакс следует принимать по 25 мг 1 раз в сутки перед сном в течение 1 нед. Затем дозу повышают с интервалом в 1-2 нед. на 25-50 мг/сут. Рекомендуемая доза составляет 100 мг/сут, максимальная суточная – 500 мг. При политерапии у детей старше 2 лет рекомендуемая суммарная суточная доза топамакса составляет от 5 до 9 мг/кг в 2 приема. Подбор дозы начинают с 25 мг/сут (или менее – 1-3 мг/кг в сутки), препарат принимают на ночь в течение 1 нед. В дальнейшем с 1-2-недель-ным интервалом дозу можно увеличить на 1-3 мг/кг в 2 приема. У подростков начальная доза топамакса при политерапии 50 мг 1 раз в сутки на ночь в течение 1 нед. Далее следует увеличивать дозу на 50-100 мг каждую неделю до подбора эффективной дозы. Средняя суточная доза составляет 200-600 мг, кратность приема – 2 раза в сутки.
В терапии затылочной эпилепсии нет необходимости в длительном медикаментозном лечении. Ребенок нуждается в курсе лечения у психотерапевта, потому что клиническая симптоматика заболевания без своевременного лечения может сделать ребенка социально неприспособленным. Прогноз лечения данной формы эпилепсии благоприятный.
В профилактических целях доктора рекомендуют родителям следить за тем, чтобы ребенок не переедал, соблюдал молочно-растительную диету и режим дня. Детям, больным затылочной эпилепсией запрещается находиться на большой высоте и заниматься туризмом, подыматься в горы, пребывать у огня или в душных помещениях, у движущихся механизмов. При этом детям показаны прогулки на свежем воздухе и легкие посильные физические нагрузки, дети должны соблюдать режим труда и отдыха.
Родителям стоит побеспокоиться о спокойной семейной атмосфере, дети с затылочной эпилепсией должны избегать стрессовых ситуаций (в детском саду, школе, дома).
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Затылочной эпилепсии у детей, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику: Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Если Вас интересуют еще какие-нибудь виды болезней и группы заболеваний человека или у Вас есть какие-либо другие вопросы и предложения – напишите нам, мы обязательно постараемся Вам помочь.www.eurolab.ua
Затылочная эпилепсия
Затылочная, или окципитальная эпилепсия (от латинского «epilepsiaoccipitalis») является гетерогенным заболеванием, которое проявляется главным образом эпилептическими припадками фокального характера, сопровождаемыми зрительными галлюцинациями.
Причины затылочной эпилепсии
Эпилептогеннные очаги размещаются в затылочной доле головного мозга из-за их первичного поражения. Заболеть затылочной эпилепсией может человек любого возраста, однако у именно у детей (в старшем возрасте) развивается идиопатическая затылочная эпилепсия.
Что касается пола, то заболевание поражает как женщин, так и мужчин. Окципитальная эпилепсия бывает наследственным заболеванием, также она может развиться после перенесённой конституциональной эпипредиспозиции.
Затылочная эпилепсия составляет 5 – 10% случаев заболевания эпилепсией, в хирургической практике доля примерно такая же.
Затылочная эпилепсия, описание и виды
Этиология затылочной эпилепсии может быть симптоматической, криптогенной, идиопатической или метаболической.
Болезнь, как правило, развивается вследствие мальформации коркового развития, а также сосудистых, неопластических, метаболических, врожденных, наследственных, воспалительных, инфекционных поражений.
Затылочные доли мозга поражаются метаболическими или другими нарушениями. Выявлена связь между затылочной эпилепсией и целиакией (глютеновойэнтеропатией) –мультифакториальным заболеванием, вызывающим нарушение пищеварения, и затылочной эпилепсией.
Помимо прочего, приступы рассматриваемой эпилепсии бывают первыми клиническими проявлениями митохондриальных нарушений (вид наследственного заболевания), или болезни Лафора – редкого генетического заболевания.
К клиническим проявлениям затылочной эпилепсии относят, прежде всего, окуломоторные и зрительные симптомы. Зрительные симптомы проявляются в элементарных и (реже) сложных зрительных галлюцинаций; слепоте; зрительных иллюзиях и палинопсии (зрительной персеверации).
К окуломоторным симптомам относятся частые непроизвольные колебательные движения глаз, их тоническая девиация и повторяющиеся моргания или трепетания век.
Галлюцинации при затылочной эпилепсии обычно представляют собой быстро развивающийся (за секунды) разноцветный круговой паттерн, который держится несколько мгновений, в редких случаях – продолжается от 1 до 3 минут.
Кроме того, спутниками затылочной эпилепсии могут стать субъективные ощущения в области глазных яблок у заболевшего, пароксизмальные нарушения полей зрения и девиация головы и шеи.
Приступы при затылочной эпилепсии
Приступы затылочной эпилепсии могут передаваться передним отделам и продуцировать симптомы, которые характерны для других долей мозга – височной, лобной или теменной. Ещё случаются генерализованные тонико-клонические приступы или вторичные гемиконвульсии. Головная боль, наступающая после припадка, очень похожа на мигрень, но возникает только у половины или у трети заболевших. Приступы наступают часто, бывает, несколько раз в день, чаще всего наступают в светлое время суток. Приступы могут группироваться в кластеры.
Диагностика и лечение заболевания
Для верной диагностики необходимо МРТ – исследование, реже могут понадобиться биохимия (для метаболических нарушений), анализ крови и ДНК, а в особых случаях–биопсия тканей, например, кожи.
В интериктальной ЭЭГ при симптоматических формах наблюдаются следующие симптомы: нарушение фоновой ритмики, проявляющееся латерализованным замедлением в задних отделах, реакции усвоения ритма фотостимуляции асимметрия альфа, часто – унилатеральные затылочные спайки. Фоновая ритмика – без нарушений, в затылочных отделах иногда могут образоваться спайки или другие пароксизмальные феномены
Иктальная ЭЭГ выявляет затылочную пароксизмальную быструю активность и спайки; которым иногда предшествует кратковременное уплощение (электродекремент) в задних отделах. В 30% случаях, однако, иктальная ЭЭГ неспособна выявить симптомы затылочной эпилепсии.
Прогноз относительно частоты повторов приступов, их тяжести и ответа на терапию варьируется от благоприятного до некурабельности приступов. В зависимости от этиологии, течение болезни может быть прогрессирующим.
Дифференциальная диагностика выявляет психогенные приступы и мигрень. Зрительная аура, возникающая при мигрени, по ряду признаков отличается от эпилептических пристопов до зрительных приступов.
Лечение затылочной эпилепсии бывает медикаментозным или хирургическим, но работа хирурга требуется лишь в тяжёлых случаях. Медикаменты способны эффективно вылечить фокальные приступы. Препараты первого выбора – это«Карбамазепин», «Ламотриджин» и «Леветирацетам». Если у ребёнка затылочная эпилепсия, то его следует показать неврологу и психотерапевту.
epi-lepsy.ru
Как проявляется затылочная эпилепсия?
Эпилепсия — это заболевание, при котором возникает повышенная элекроактивность участка головного мозга с развитием определенного рода припадков. Характер их зависит от локализации процесса.
Затылочная эпилепсия, или окципитальная, относится к гетерогенным болезням, при которых у больного возникают фокальные приступы с галлюцинациями зрительного характера.
Почему возникает?
Причиной развития этого вида эпилепсии является первичное поражение затылочной области мозга. Развивается она у людей любой возрастной группы. Идиопатическая затылочная эпилепсия бывает в детском возрасте. Поражает этот недуг и женщин, и мужчин. Встречаются и случаи проявление заболевания в результате наследственной предрасположенности.
Окципитальная эпилепсия, согласно данным статистики, составляет около10% от всех случаев зарегистрированной эпилепсии.
Затылочные участки поражаются в результате:
- Заболевания сосудов мозга.
- Метаболических нарушений.
- Врожденной патологии.
- Отягощенной наследственности.
- Воспалительных и инфекционных процессов с поражением мозга.
Существует взаимосвязь между целиакией и данным заболеванием. Иногда оно развивается на фоне болезни Лафора, которая относится к редким наследственным заболеваниям
Виды заболевания
Затылочная эпилепсия бывает:
- симптоматическая;
- криптогенная;
- идиопатическая;
- метаболическая.
У детей
Затылочная эпилепсия у детей может развиваться как в раннем (3−6 лет), так и в более позднем возрасте (6−14 лет). Это так называемая доброкачественная эпилепсия с развитием простых парциальных приступов и зрительных расстройств.
Для данного заболевания характерным признаком является развитие припадка во сне или сразу после пробуждения. Нельзя исключить вторичные генерализованные приступы при распространении возбуждения на передние части мозга. Иногда они сопровождаются тошнотой и рвотой.
У маленьких детей приступы имеют тяжелое течение и могут длиться от нескольких часов до нескольких дней.
Симптомы
Основными признаками этой болезни являются простые, но бывают и сложные зрительные галлюцинации. Иногда развивается слепота или потеря зрения не определенный промежуток времени. Больной видит черноту или белую пелену. Часто приступы сочетаются с мигренеподобной болью в голове, которая отмечается у половины пациентов с этим диагнозом.
Окуломоторные признаки выражаются в колебании глаз, развивается тоническая девиация глаз и головы в противоположных направлениях, частые моргания или трепетание век по типу крыльев бабочки.
Больной видит световые вспышки или пятна, звездочки, квадраты или полосы. Они могут быть цветными, перемещаться горизонтально, кружится, перемещаться ближе и отдаляться.
Приступы развиваются без изменения сознания. Начальные признаки характеризуются только симптоматикой поражения окципитальной части мозга, следом могут присоединяться и другие симптомы, которые связаны с распространением возбуждения на прочие участки мозга.
При развитии подобной симптоматики и с целью грамотного купирования приступа следует обращаться к специалисту.
Диагностика
Для подтверждения диагноза используется ЭЭГ с использованием поверхностных электродов. Ее проводят в период между приступами. Это исследование очень информативно подтверждает этот диагноз в виде высокоамплитудных пиков активности в затылочных отведениях. Морфологически паттерны немного напоминают роландический вид эпилепсии.
Для этого заболевания характерным признаком является исчезновение активности при исследовании с открытыми глазами.
При метаболических нарушениях проводятся лабораторные биохимические анализы. При подозрении на наследственную патологию используется анализ ДНК. Если есть необходимость, то врач рекомендует проведение биопсии кожи.
При необходимости исключения патологии в головном мозге проводится МРТ.
Исследование внимания, нарушений памяти и поведения, отклонений зрительного характера проводится нейропсихологическое исследование.
После использования всех этих методик на основе полученных данных ставится диагноз.
Лечение и терапия
Лечение назначается в том случае, если приступы имеют частый характер, или заболевание сильно снижает качество жизни пациента. У детей при этом может нарушаться психосоциальное поведение.
Если приступы не имеют генерализованного характера, то препаратами выбора являются карбамазепины или вальпроаты. При их непереносимости или неэффективности применяется новое средство для лечения — топирамат. Его можно использовать у детей, которым исполнилось 2 года. В качестве резервного средства выступает фенитоин.
Особенностями лечения затылочной эпилепсии является то, что в детском возрасте не существует необходимости применения лекарственных средств непрерывно. В комплексном лечении должен участвовать психотерапевт, для того, чтобы помочь ребенку социально адаптироваться.
Прогноз при лечении затылочной формы эпилепсии, чаще всего, благоприятный. Ремиссия после лечения составляет около 95%. Но процесс купирования приступов гораздо сложнее, и дозировка необходимых средств несколько выше. Как правило, фокальные приступы удается купировать медикаментозными средствами.
Хирургические методы используются только в самых крайних случаях.
Профилактика
Для предупреждения развития затылочной эпилепсии в детском возрасте родители должны следить за тем, чтобы ребенок нормально питался, не переедал. В рационе должно быть много растительной и молочной пищи.
Не рекомендуется людям с таким диагнозом подниматься на высоту или заниматься туристическими видами спорта. Пациенту нужен свежий воздух, ему не показано пребывание в плохо проветриваемом помещении или около открытого огня.
Обязательно должен соблюдаться режим отдыха с легкими физическими нагрузками. Детям необходимо нормальная и уютная семейная атмосфера.
Оцените статью: (голосов: 1 , среднее: 5,00 из 5) Loading ...
vashnevrolog.ru
Затылочная эпилепсия у детей и взрослых: симптомы, причины, лечение
Затылочная эпилепсия у взрослых и детей – это форма неврологического заболевания, при которой эпилептогенный очаг локализируется в затылочных долях. Она может развиваться как у мужчин, так и у женщин, ее доля среди всех больных этим недугом составляет 5-10%. Заболевание может возникнуть в любом возрасте. Исключением является идиопатическая форма патологии, которая развивается преимущественно у детей. Основу лечения затылочной эпилепсии составляет медикаментозная терапия, однако согласно статистическим данным в 10% случаев требуется хирургическое вмешательство.
Виды затылочной эпилепсии
В зависимости от причины происхождения затылочная эпилепсия бывает:
- Симптоматическая затылочная эпилепсия – возникает на фоне врожденных, приобретенных и прогрессирующих поражений тканей мозга. Причинами ее развития могут служить черепно-мозговые травмы и опухоли в затылочной части головы, воспалительные, инфекционные и сосудистые заболевания, а также патологии, полученные в утробе матери или во время родовой деятельности.
- Идиопатическая затылочная эпилепсия – форма заболевания, развивающаяся на фоне наследственной предрасположенности и генетики. Идиопатическая эпилепсия носит преимущественно доброкачественный характер, проявляется в детском возрасте. Наиболее распространенной разновидностью этой формы является затылочная эпилепсия по типу Гасто.
- Криптогенная затылочная эпилепсия – патология с невыясненным характером происхождения. Симптомы и диагностические исследования свидетельствуют о чрезмерном возбуждении нервных клеток именно в этой части головы, при этом какие-либо наследственные или патологические причины ее происхождения отсутствуют.
- Метаболическая затылочная эпилепсия – возникает на фоне врожденных нарушений обмена веществ, например, эклампсии.
Приступы затылочной эпилепсии отличаются молниеносным развитием и кратковременностью. Их длительность обычно варьируется от нескольких секунд до 3-х минут. Припадки могут развиваться по несколько раз в день в период бодрствования и активной деятельности, при этом группироваться в кластеры.
Основными клиническими проявлениями затылочной эпилепсии являются:
- Сохранность сознания, за исключением случаев, когда эпилептический разряд распространяется на другие отделы головного мозга.
- Зрительные расстройства – в момент приступа у больного могут возникать галлюцинации, зрительные иллюзии, выпадение полей зрения, глазная боль, нистагм, дрожание и частое моргание век, кратковременная слепота. При простых галлюцинациях у больного перед глазами возникают разноцветные круги, молниеносно увеличивающиеся в размерах. Сложные зрительные иллюзии появляются при распространении электрического разряда на передние отделы затылочной части, что довольно часто также приводит к появлению слепоты и развитию генерализированных судорожных припадков.
- Головная боль, по характеру схожая с мигренью, возникает сразу после приступа у 30-50% больных.
- Тонико-клонические судороги – появляются при переходе фокальной затылочной эпилепсии во вторично-генерализированный тип.
- Вегетативные признаки – повышение температуры тела, бледность кожи, вялость, слабость, приступы раздражительности, затрудненность дыхания, недержание кала, а также непроизвольное мочеиспускание, в большинстве случаев заканчивающееся рвотой.
При распространении повышенного электрического разряда с затылочной части на передние отделы головного мозга у больного могут проявиться признаки теменной эпилепсии. В таком случае у больного развивается затылочно-теменная эпилепсия. При распространении патологического нервного импульса на височную часть у пациента диагностируется височно-затылочная эпилепсия.
Затылочная эпилепсия у детей
Заболевание этой формы может проявиться как у взрослых, так и у детей. Наиболее часто в детском возрасте проявляются два основных синдрома затылочной доброкачественной эпилепсии – Панайотопулоса и Гасто.
Синдром Панайотопулоса характеризуется ранним началом – в 75% случаев его дебютирование приходится на 3-6 лет. Заболевание крайне редко носит наследственный характер, при этом вероятность его развития в результате сложной родовой деятельности предельно высока. В 17% случаев у больных проявляются приступы фебрильных судорог, при тяжелых состояниях наблюдается потеря сознания с высоким риском перехода приступа в тонико-клонический генерализированный припадок. Синдром Панайотопулоса отличается одним из самых благоприятных прогнозов среди всех форм этого заболевания. В 30% случаев припадки проявляются однократно и лишь у 10% детей их количество превышает 10 приступов. При правильно подобранной медикаментозной терапии уже через 1-2 года можно добиться полной ремиссии.
Синдром по типу Гасто проявляется в более позднем возрастном периоде – от 6-ти до 14-ти лет. Заболевание относят к идиопатической форме затылочной эпилепсии, так как причиной его возникновения является генетическая составляющая. Основными симптомами синдрома Гасто являются зрительные расстройства, при этом сознание в момент приступа остается сохранным. Обязательным условием в лечении патологии является прием противосудорожного средства, что позволяет в большинстве случаев уже через 2-4 года добиться прекращения приступов.
В любом возрасте у детей может развиться симптоматическая затылочная эпилепсия. Причинами ее возникновения в таком случаев являются внутриутробные заболевания, а также патологии, полученные во время и после рождения.
Лечение
В большинстве случаев для лечения затылочной эпилепсии, как и для остальных форм фокальной эпилепсии, назначаются медикаментозные препараты. В тяжелых случаях больному может быть проведено хирургическое вмешательство.
Противоэпилептическими средствами первой очереди, назначаемыми при затылочной эпилепсии, являются Карбамазепин, Ламотриджин и Леветирацетам. Один из препаратов назначается в минимальной дозировке, которая на протяжении 7-ми дней доводится до стандартной суточной дозы. В случае отсутствия эффекта от лечения в течение 2-3-х месяцев препарат может быть заменен или дополнен другим.
При синдроме Панайотопулоса, развивающемся у детей, принцип лечения будет зависеть от частоты и продолжительности приступов. Однократные и короткие припадки довольно часто не требуют противоэпилептической терапии. При частых и регулярно повторяющихся приступах ребенку в зависимости от его возраста может быть назначен Карбамазепин, Сультиам или Клобазам.
В случае развития тяжелых приступов затылочной эпилепсии больному необходимо оказать неотложную медицинскую помощь, для чего вызвать бригаду скорой помощи.
Прогноз
Прогноз затылочной эпилепсии зависит от формы заболевания, степени тяжести поражения нервных клеток, а также индивидуальных особенностей организма.
При симптоматической затылочной эпилепсии шансы на снижение частоты и интенсивности припадков возрастают при условии регулярного приема медикаментозных препаратов и устранении первопричины заболевания. В противном случае не исключены случаи прогрессирования недуга и ухудшения состояния.
При доброкачественной затылочной эпилепсии прогноз на выздоровление относительно благоприятный. У 50% больных при условии приема противоэпилептических препаратов приступы исчезают через 3-4 года после начала заболевания. В остальных случаях, особенно при отсутствии медикаментозной терапии, приступы могут сохраняться, сопровождаясь при этом зрительными расстройствами.
При выявлении доброкачественной затылочной эпилепсии у детей в раннем возрасте прогноз весьма благоприятный. В таких случаях полной ремиссии удается достичь в течение 1-2-х лет.
Автор: Иван Дроздов, невролог
Информация на сайте создается для тех, кому необходим квалифицированный специалист, не нарушая привычный ритм собственной жизни.
progolovy.ru
Затылочные эпилепсии у детей
Затылочные эпилепсии у детей
Н.А. Ермоленко, М.В. Уварова, И.А. Бучнева, Е.А. Балакирева, А.Ю. Ермаков
Childhood occipital epilepsy
N.A. Ermolenko, M.V. Uvarova, I.A. Buchneva, E.A. Balakireva, A.Yu. Ermakov
Воронежская государственная медицинская академия им. Н.Н. Бурденко;
Воронежская областная детская клиническая больница №1; Московский НИИ педиатрии и детской хирургии
Затылочные фокальные эпилепсии — полиморфная полиэтиологическая группа заболеваний, составляющая приблизительно 5—10% всех эпилепсий. Затылочные эпилепсии характеризуются простыми парциальными и вторично генерализованными приступами и могут быть идиопатическими, симптоматическими или предположительно симптоматическими. Симптоматические затылочные эпилепсии могут начинаться в любом возрасте, идиопатические — возникают исключительно в детском возрасте. Трудности дифференциальной диагностики затылочных эпилепсий у детей обусловлены незрелостью детского мозга и преобладанием в клинической картине автономных признаков. Прогноз и лечение зависят от этиологии затылочных эпилепсий.
Ключевые слова: дети, затылочные эпилепсии, противоэпилептические препараты.
Occipital focal epilepsies are a polymorphic polyetiological group of diseases, which accounts for about 5—10% of all epilepsies. Occipital epilepsies are characterized by simple partial and secondarily generalized seizures and they may be idiopathic, symptomatic, or presumably symptomatic. Symptomatic occipital epilepsies may occur at any age; idiopathic ones solely in childhood. Difficulties in the differential diagnosis of childhood occipital epilepsies arise from immaturity of the child's brain and a preponderance of autonomic signs in the clinical picture. The etiology of occipital epilepsies determines their prognosis and treatment.
Key words: children, occipital epilepsies, antiepileptic drugs.
Затылочные эпилепсии характеризуются эпилептическими приступами вследствие разрядов из затылочного фокуса, возникающих спонтанно или под воздействием внешних зрительных раздражителей [1—5].
Такие эпилепсии могут быть идиопатически-ми, симптоматическими или предположительно симптоматическими [6]. Комиссией по классификации и терминологии Международной лиги по борьбе с эпилепсией [7] затылочные эпилепсии отнесены к локально-обусловленным эпилепси-ям и эпилептическим синдромам и описываются как «эпилептические синдромы, обычно характеризующиеся простыми парциальными и вторично генерализованными приступами». Появление сложных парциальных приступов обусловлено распространением возбуждения из затылочных долей в другие области мозга. Вопрос частоты ассоциации затылочных приступов и мигрени сложный и спорный.
В новой диагностической схеме ILAE [8] затылочные эпилепсии еще не детализированы.
© Коллектив авторов, 2009
Ros Vestn Perinatol Pediat 2009; 1:37-44
Адрес для корреспонденции: 125412 Москва, ул. Талдомская, д. 2
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.Эпидемиология
Затылочные эпилепсии составляют приблизительно 5—10% всех эпилепсий [9—11]. Симптоматические затылочные эпилепсии могут начинаться в любом возрасте, на любой стадии во время или после состояний, послуживших причиной возникновения эпилептических приступов. Идиопатиче-ские эпилепсии возникают исключительно в детском возрасте [6].
Этиология
Возникновение симптоматических затылочных эпилепсий связано с наличием структурных дефектов или метаболических нарушений [6]. У больных с симптоматическими затылочными эпилепсиями структурные изменения в затылочных долях могут быть врожденными, резидуальными, а также прогрессирующими вследствие сосудистой, опухолевой, наследственной, воспалительной, паразитарной, инфекционной или системной патологии. Нарушения кортикального развития являются наиболее частой причиной симптоматических затылочных эпилепсий [9, 12].
Метаболические заболевания или другая патология (эклампсия) могут быть причиной возникновения затылочных эпилептических приступов, однако в этих случаях диагноз эпилепсии не ставится. Известны случаи ассоциации целиакии с
затылочной эпилепсией, вызванной кальцинатами в затылочной области [13, 14]. Затылочные приступы могут наблюдаться в дебюте таких дегенеративных заболеваний, как болезнь Лафора [15, 16], или митохондриальной патологии [17].
Патофизиология
Простые зрительные галлюцинации возникают вследствие эпилептических разрядов, которые генерируются в первичной зрительной коре, сложные зрительные галлюцинации — в зоне стыка затылочно-теменно-височных долей, зрительные иллюзии — в субдоминантном полушарии теменной доли [18]. Разряды могут распространяться из затылочной доли в другие регионы мозга, продуцируя симптомы, характерные для височной, теменной и лобной долей, а также гемиклонические приступы и вторично-генерализованные тонико-клонические судороги. Очень часто в момент приступа и в постприступном периоде развивается головная боль [6].
Частая ассоциация окципитальных эпилептических приступов с головной болью предполагает тесную взаимосвязь затылочной доли и тригеми-новаскулярной системы или стволовых структур, ответственных за мигренозную головную боль. Таким образом, эпилептические приступы генерируют мигренеподобные атаки [4,19].
Клиническая манифестация
Иктальные (приступные) клинические проявления затылочной эпилепсии характеризуются субъективными и объективными симптомами, которые в большинстве случаев являются зрительными и глазодвигательными [6].
Зрительные симптомы:
— простые и реже сложные зрительные галлюцинации;
— зрительные иллюзии;
— пароксизмальный амавроз;
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.— пароксизмальные нарушения полей зрения;
— сенсорные галлюцинации в виде ощущения движений глазных яблок;
— боль в глазных яблоках.
Объективные глазодвигательные симптомы:
— тоническая девиация головы и глаз;
— окулоклонические движения или нистагм;
— моргания или трепетание век.
Простые зрительные галлюцинации наблюдаются у 47—73% больных с затылочными эпилепси-ями [2, 20]. Как правило, это первый и иногда единственный симптом начинающегося затылочного приступа. Подробное описание простых зрительных галлюцинаций при идиопатических и симптоматических эпилепсиях дал С Panayiotopoulos [6].
Цвет, форма и размер. Преимущественно паттерны цветные, как правило, разноцветные (превалируют красный, желтый, голубой и зеленый цвета) в виде кругов, пятен, шаров, обычно множественных, и очень редко единичных. Цветные зигзаги, треугольники, прямоугольники или звезды единичные или в комбинации с кольцевидными паттернами встречаются значительно реже. Вспышки, блики бесцветных огней, наоборот затемнения или нециркулярные паттерны — крайне редки.
Локализация. Локализуются вначале унилате-рально, чаще в височных полях зрения. Расположение их в центре поля зрения или неопределенная локализация встречается лишь у 10—30% пациентов [6, 21].
Движения. Компоненты простых зрительных галлюцинаций, как правило, множественные, могут расширяться без каких-то определенных движений. Движения по направлению к центру поля зрения или в противоположную сторону возникают значительно реже. Крайне редко отмечаются движения вокруг оси, по кругу, ротаторные движения, ощущение приближения и удаления [21—23].
Латерализация. Простые зрительные галлюцинации возникают на стороне, противоположной очагу эпилептогенеза. Двигающаяся горизонтально по направлению к противоположной стороне унилатеральная галлюцинация возникает ипсила-терально эпилептогенному очагу [24].
Зрение. Во время приступа зрение может быть нарушено. Расплывчатость, нечеткость, смазан-ность изображения могут появиться в начале зрительных галлюцинаций или как самостоятельный эпилептический приступ [6].
Продолжительность. Простые зрительные галлюцинации обычно короткие, продолжительностью от нескольких секунд до 1—3 мин, реже дольше [4, 25—27]. Исключительно редко продолжительность составляет более 20—50 мин, характеризуя фокальный зрительный эпилептический статус без других судорожных симптомов [18, 28].
Частота и циркадность. Зрительные приступы возникают в виде множественных кластеров ежедневно или раз в неделю, обычно в ночное время или при пробуждении [6].
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.Стереотипность появления. Иктальные симптомы простых зрительных галлюцинаций стереотипны в своих проявлениях у одного и того же пациента, исключая продолжительность приступа. Крайне редко у одного пациента могут возникать различные типы приступов [6].
Сложные зрительные галлюцинации могут принимать формы людей, животных, объектов, фигур или сцен. Могут быть знакомыми (хорошо известными) или незнакомыми, дружелюбными
или враждебными, простыми или вычурными. Они могут занимать незначительную или большую часть половины поля зрения, находиться в центре или занимать все поле зрения. Могут быть неподвижными или двигаться горизонтально, расширяться или сужаться, приближаться или удаляться. У пациентов со структурным дефектом головного мозга сложные зрительные галлюцинации появляются в зрительном поле на стороне очага. Сложные зрительные галлюцинации никогда не имеют эмоциональный или сложный характер, как при височных приступах [4, 26, 29].
Зрительные иллюзии возникают как ошибочная интерпретация, ложные ощущения реально существующих изображений или предметов. Эти нарушения восприятия включают изменение размера, величины, формы, пропорций, положения, цвета, освещения и движения. Иктальные зрительные иллюзии могут занимать часть или все поле зрения и более характерны для симптоматических затылочных эпилепсий, чем для идиопатических.
Сложные зрительные галлюцинации, зрительные иллюзии и другие симптомы при распространении эпилептического разряда в передние отделы могут заканчиваться гемиконвульсиями и генерализованными конвульсиями [6]. Иногда они служат первым клиническим проявлением эпилептического приступа, но чаще следуют за простыми зрительными галлюцинациями.
Сенсорные галлюцинации в виде ощущения движений глазных яблок в отсутствие видимых движений встречаются редко и являются типичными для затылочной эпилепсии [23, 30].
Иктальный амавроз проявляется в виде нечеткости или врёменной утраты зрения. Больные обычно описывают свои ощущения как «черноту перед глазами», реже как «белую пелену» [2]. Пароксиз-мальный амавроз нередко сочетается с простыми зрительными галлюцинациями, но чаще всего возникает в начале и является единственным признаком эпилептического приступа с внезапным развитием [4, 11, 12, 26, 31]. Пароксизмальный амавроз обычно продолжительнее (3—5 мин) зрительных галлюцинаций. Описаны случаи пароксизмально-го амавроза в течение часов или несколько дней, которые представляют собой вариант эпилептического статуса (status epilepticus amauroticus) [32].
Тоническая девиация головы и глаз — частый, но не обязательный признак, характерный для эпилептических пароксизмов различной локализации. При затылочной эпилепсии девиация головы и глаз нередко является одним из манифестных симптомов приступа, при этом голова и глаза поворачиваются в контралатеральную эпилептическому фокусу сторону [10]. Сознание часто, но не всегда нарушено.
Иктальный нистагм — как правило, горизонтальный или редко вертикальный. Быстрая фаза нистагма противоположна эпилептическому фокусу [6].
Моргание, трепетание век имеет насильственный характер и напоминает трепетание крыльев бабочки [2]. Этот симптом возникает обычно после фазы зрительных галлюцинаций и является предвестником вторично генерализованных судорог. Тем не менее он может появиться в самом начале приступа или быть его единственным признаком. Иногда трепетание век бывает настолько незаметным, что не расценивается как эпилептический приступ, и обнаруживается только при записи ЭЭГ у пациентов с затылочной фотосенситивностью [6].
Симптом насильственного моргания или трепетания век может встречаться при различных типах эпилептических пароксизмов, поэтому его диагностическая значимость должна оцениваться с осторожностью [10].
Симптом «широко открытых глаз» подробно описан при затылочных эпилепсиях, но может встречаться при других локализациях эпилептического фокуса. Так, широко раскрытые глаза с фиксацией взгляда и расширением зрачков (staring) — типичный симптом мезиальной височной эпилепсии [33].
Сознание во время простых и сложных зрительных галлюцинаций, амавроза и других субъективных затылочных приступов, как правило, сохранено, но может нарушаться или полностью утрачиваться во время приступов с девиацией глазных яблок, морганиями и конвульсиями.
Иктальная головная боль часто связана с затылочными эпилептическими приступами [4, 11, 26, 31]. Она в основном локализована в области орбит (ретроорбитальная), как правило, внезапная, острая, сопровождается чувством сдавления висков, неопределенной болью в голове или чувством электрических прострелов.
Постприступная головная боль, часто неотличимая от мигрени, наиболее характерна для затылочных эпилепсий (встречается более чем в 50% случаев). Боль может возникать даже после очень коротких зрительных пароксизмов [4, 26, 34, 35], как правило, появляется на 3—15-й минуте после окончания зрительного приступа [36].
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.Постиктальное нарушение полей зрения встречается относительно редко и проявляется в виде пароксизмальной гемианопсии (квадрантной ге-мианопсии) продолжительностью от нескольких секунд до нескольких минут [10, 18].
Распространение эпилептического разряда. Эпилептические разряды могут распространяться из затылочной доли в другие регионы мозга, продуцируя симптомы височной, теменной и лобных
долей, а также гемиклонические и вторично генерализованные судороги. Эпилептогенный фокус из нижней части шпорной извилины генерирует разряды, распространяющиеся в височную долю, вызывая сложные парциальные приступы. Разряды из верхней части шпорной извилины распространяются в теменную и лобную доли, вызывая преимущественно моторные приступы.
Присоединение височно-долевой симптоматики практически не встречается при идиопатиче-ских затылочных эпилепсиях [6].
Диагностика
Проведение магнитно-резонансной томографии (МРТ) высокого разрешения необходимо во всех случаях затылочных эпилепсий [4, 11, 26, 31]. В редких случаях сочетания целиакии и затылочных эпилептических приступов требуется проведение компьютерной томографии для идентификации кальцинатов в головном мозге. В случаях преоперационной подготовки необходимо проведение функциональной МРТ [37].
Некоторые случаи симптоматических затылочных эпилепсий требуют биохимического скрининга, ДНК-анализа и даже биопсии кожи или других тканей для исключения метаболического заболевания [18]. В дебюте болезни Лафора генерализованные тонико-клонические припадки приблизительно у половины больных сочетаются с парциальными затылочными пароксизмами, проявляющимися зрительными галлюцинациями, скотомами, преходящей корковой слепотой. Болезнь Лафора — наследственное аутосомно-рецессивное заболевание, специфическим морфологическим признаком которого является наличие в ЦНС особых интранейрональных включений — амилопек-тиноподобных полисахаридов (телец Лафора), что дает основание отнести данную патологию к болезням накопления. Основным лабораторным методом, подтверждающим прижизненный диагноз болезни Лафора, является световая микроскопия биоптатов кожи, позволяющая обнаружить тельца Лафора в клетках протоков потовых желез.
Электроэнцефалография с использованием поверхностных электродов — неотъемлемая часть обследования больных. Но этот метод имеет существенные ограничения и может быть недостаточно информативным [23, 30].
Интериктальная электроэнцефалограмма (ЭЭГ). В симптоматических случаях на фоновой ЭЭГ обычно выявляются медленные волны в задних отведениях. Кроме того, могут быть унилатераль-ные затылочные спайки, быстрые полиспайки и иногда билатерально синхронизированные разряды в затылочных отведениях. Может выявляться затылочная фотосенситивность [18]. Ухудшение
фоновой ЭЭГ указывает на прогрессирование заболевания.
В идиопатических случаях фоновая интериктальная ЭЭГ — нормальная [18]. Регистрируются спонтанные и/или вызванные окципитальные спайки и окципитальные пароксизмы, которые исчезают с возрастом, однако гораздо позже, чем прекращаются эпилептические приступы.
Иктальная ЭЭГ. Иктальные паттерны обычно начинаются с пароксизмальной быстрой активности, быстрых спайков, локализованных в затылочных отведениях с распространением в некоторых случаях на передние отделы и генерализацией в виде нерегулярных спайк-волновых разрядов или мономорфной спайк-волновой активности. Короткие затылочные вспышки могут регистрироваться до появления быстрой ритмичной активности. Часто у пациентов с симптоматической затылочной эпилепсией иктальные разряды регистрируются не только в затылочных отведениях. Обычно отсутствует постикальное региональное замедление за исключением затяжных и вторично генерализованных приступов. У одной трети пациентов иктальная поверхностная ЭЭГ не обнаруживает характерных для затылочной локализации изменений [18].
Дифференциальная диагностика
Затылочные эпилептические приступы должны прежде всего дифференцироваться с мигренью, нормальными двигательными феноменами, психогенными и другими неэпилептическими пароксизмами [18]. Необходимо дифференцировать симптоматические и идиопатические затылочные эпилепсии ввиду различного прогноза и подходов к лечению.
Зрительные приступы при симптоматических и идиопатических затылочных эпилепсиях неразличимы [4]. В симптоматических случаях ок-ципитальные разряды чаще распространяются в соседние регионы и приобретают черты экстраок-ципитальных приступов, особенно височных. Отсутствие изменений неврологического статуса и нормальные результаты нейровизуализации головного мозга могут ошибочно привести к установлению диагноза идиопатической формы, поэтому рекомендуется проведение высокоразрешающей МРТ с использованием сканеров нового поколения [12, 18].
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.Прогноз
Частота, тяжесть эпилептических приступов, а также результаты лечения зависят от конкретного случая и степени повреждения головного мозга [18].
Симптоматические затылочные эпилепсии
Причинами возникновения симптоматических затылочных эпилепсий являются дисгенезии мозга (фокальная корковая дисплазия, микрогирия и др.), опухоли, нейрофиброматоз, кисты в затылочной доле, врожденные пороки развития мозга, нейроинфекции [10, 23]. Затылочные эпилепсии характеризуются преимущественно простыми парциальными пароксизмами, не сопровождающимися нарушением сознания. Клинические проявления затылочных эпилепсий разделяются на ранние и поздние [2]. Ранние симптомы обусловлены эпилептической активностью непосредственно в затылочной доле, тогда как поздние являются результатом распространения эпилептической активности на другие области мозга [23, 30].
При вовлечении в процесс медиальных отделов височной доли появляются типичные признаки медиальной височной эпилепсии — эпигастраль-ная аура, ороалиментарные и кистевые автоматизмы. Если начальные симптомы затылочных приступов своевременно не распознаются, больному устанавливается ошибочный диагноз височной эпилепсии [2]. Симптомами распространения эпилептической активности на латеральные отделы височной доли являются сложные слуховые (музыка, хор голосов) и зрительные галлюцинации (картины и сцены с образами животных, людей), зрительные иллюзии (нарушения формы и величины предметов, расстройства восприятия пространства, ощущения убыстрения или замедления движения предметов). Тонические позы, наблюдаемые при вовлечении латеральной (сильвиевой) борозды, вероятно, свидетельствуют и об активации эпилептическими разрядами дополнительной моторной зоны [2]. Эпилептическое головокружение может быть как проявлением затылочных пароксизмов (поле 19 по Бродману), так и признаком распространения эпилептической активности на теменно-височную область [10].
Идиопатические затылочные эпилепсии
Детская затылочная эпилепсия с поздним дебютом (тип Гасто) характеризуется приступами, протекающими преимущественно в виде пароксизмов зрительных нарушений и нередко заканчивающимися мигренозной головной болью. На ЭЭГ отмечаются ритмичные комплексы «спайк—вол-на» в затылочных областях, возникающие при закрывании глаз. Впервые эта форма была описана H. Gastaut в 1950 г. как «доброкачественная затылочная эпилепсия детского возраста», однако слово «доброкачественная» из названия было убрано, так как в отличие от роландической эпилепсии не все пациенты полностью выздоравливали в подростковом возрасте. Лишь спустя 40 лет после
первого сообщения было предложено название «детская затылочная эпилепсия с поздним дебютом (тип Гасто)», чтобы отличать его от синдрома Панайотопулоса — доброкачественной детской затылочной эпилепсии с ранним дебютом, также проявляющейся затылочными пароксизмами.
Распространенность болезни составляет 0,2— 0,9% от общего числа случаев эпилепсии и 2—7% от общего числа доброкачественных фокальных эпилепсий [6]. Мальчики и девочки подвержены заболеванию в равной степени. Дебют эпилепсии — в возрасте 3—16 лет с пиком в 8 лет.
Приступы при синдроме Гасто характеризуются зрительными симптомами, которые обычно возникают первыми и являются наиболее типичными. Спектр зрительных расстройств, наблюдаемых во время приступа, весьма разнообразен и варьирует по числу различных симптомов. Так, преходящие нарушения зрения встречаются в 65% случаев, амавроз — в 52%, элементарные зрительные галлюцинации (мелькание светящихся предметов, фигур, вспышки света перед глазами) — в 50%, сложные, сценоподобные галлюцинации — в 14% [38]. Сознание, как правило, сохранено. Приступы возникают в основном в дневное время. Незрительные феномены также разнообразны: ге-миклонические судороги — в 13%, автоматизмы — в 13 %, дисфазия, дизестезия, версивныедвижения— в 25% [38]. Постприступное состояние в ряде случаев характеризуется мигренеподобной головной болью (33% случаев), иногда сопровождающейся тошнотой и рвотой (17% случаев) [38]. Одним из частых (25%) факторов, провоцирующих развитие приступа, является резкая смена освещенности при переходе из темного помещения в светлое.
Интеллект у детей с затылочной эпилепсией нормальный. Очаговые неврологические нарушения отсутствуют.
Критериями диагноза детской затылочной эпилепсии с поздним дебютом являются:
— дебют в возрасте 7—8 лет;
— частая наследственная отягощенность по эпилепсии и мигрени;
— приступы, сочетающиеся со зрительными симптомами, без потери сознания;
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.— нормальное нервно-психическое развитие;
— доброкачественное течение;
— наличие на ЭЭГ ритмичных спайков в затылочной области, появляющихся при закрывании глаз.
Электроэнцефалография. На интериктальной ЭЭГ частота основного ритма соответствует возрасту. При этом в затылочных отведениях при закрытых глазах могут регистрироваться билатерально-синхронные ритмичные спайки, полностью маскирующие альфа-ритм. Окципитальные
пароксизмы часто демонстрируют fixation-off sensitivity (чувствительность к прекращению зрительной фиксации). Некоторые пациенты могут иметь только окципитальные спайки. Центротемпораль-ная, фронтальная локализация спайков может встречаться, но значительно реже, чем при синдроме Панайотопулоса. До сих пор дискутируется вопрос, является ли фотосенситивность частью этого синдрома или нет [6]. Установлено также, что в 38% случаев затылочные спайки сочетаются с генерализованными, билатеральными комплексами «спайк—волна», «полиспайк—волна» [38]. В редких случаях может быть нормальная ЭЭГ.
Иктальная ЭЭГ характеризуется внезапным появлением окципитальных разрядов, которые состоят из быстрого ритма, быстрых спайков [39]. Они имеют гораздо меньшую амплитуду, чем ок-ципитальные пароксизмы. Как правило, постик-тальных изменений на ЭЭГ нет [6].
Доброкачественная детская затылочная эпилепсия с ранним дебютом (синдром Панайотопу-лоса) характеризуется редкими ночными пароксизмами. Это заболевание отмечается у 13% детей в возрасте 3—6 лет с одним или более афебриль-ным приступом и у 6% — в возрастной группе 1—15 лет. В общей популяции населения болезнь может быть выявлена у 2—3 детей из 1000. Эти цифры могут быть выше, если учитывать случаи атипичного синдрома Панайотопулоса.
Болезнь дебютирует в возрастном диапазоне 1—14 лет с пиком заболеваемости (76% случаев) в 4—5 лет. Мальчики и девочки болеют одинаково часто. Вегетативные припадки и вегетативный эпилептический статус — основные проявления синдрома Панайотопулоса.
Приступ, как правило, начинается с рвоты, тонической девиации глазных яблок в сторону и нарушения сознания. В ряде случаев наблюдается переход в гемиконвульсии или генерализованный тонико-клонический приступ [19, 40]. Другие вегетативные симптомы, которые появляются в начале или по мере развития приступа: бледность кожных покровов, реже — гиперемия или цианоз, мидриаз, реже — миоз, кардиореспираторные и терморегу-ляторные изменения, кашель, недержание мочи и/или кала, нарушения перистальтики кишечника, возможна гиперсаливация (более характерна для роландической эпилепсии). Головная боль наблюдается исключительно в начале приступа.
У 10% больных отмечаются только вегетативные припадки и вегетативный эпилептический статус. Они начинаются и заканчиваются вегетативными симптомами. В других случаях за вегетативными симптомами приступа следует нарушение сознания (94%), девиация глаз (60—80%), гемиконвуль-сии (26%) и генерализованные судороги (20%).
У 10—20% больных девиация глазных яблок может наблюдаться без рвоты. Около 20% приступов заканчиваются гемиконвульсиями, часто с джексо-новским маршем или генерализованными припадками. Другие сопутствующие симптомы приступа включают в себя роландические проявления, такие как остановка речи (8%), гемифациальные (6%), фарингооральные приступы (3%). Иктальные визуальные симптомы (6%), такие как зрительные галлюцинации, иллюзии или слепота, наблюдаются после возникновения более типичных для синдрома Панайотопулоса симптомов. Унилатераль-ное опущение угла рта, слабые миоклонические судороги, трепетание век, иктальный нистагм или автоматизмы наблюдаются менее чем в 3% случаев каждый. Припадки без вегетативных признаков — явление редкое (7%).
Продолжительность приступов вариабельна: от нескольких минут до нескольких часов [6]. В 44% случаев приступ длится больше 30 мин и может продолжаться до 7 ч (в среднем около 2—3 ч), являясь, таким образом, эпилептическим статусом. В остальных случаях приступы продолжаются от 1 до 30 мин (в среднем 9 мин) [6]. Нередко у больных наблюдается статус парциальных приступов [40].
Критерии диагноза. Основные критерии диагноза доброкачественной затылочной эпилепсии с ранним дебютом включают:
— дебют в возрасте 4—6 лет;
— приступы с потерей сознания, рвотой, девиацией глазных яблок и головы;
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.— преимущественно ночной характер пароксизмов;
— частые эпизоды вегетативного эпилептического статуса (до 50% всех приступов);
— нормальный интеллект и нервно-психическое развитие;
— доброкачественное течение;
— наличие на ЭЭГ спайков или комплексов острая—медленная волна в затылочных областях.
Электроэнцефалография. На интериктальной ЭЭГ частота основного ритма соответствует возрасту. У 90% пациентов обнаруживаются спайки — высокоамплитудные комплексы острая—медленная волна [6, 41], которые чаще (68% случаев) локализуются в задних отделах. У 64% больных регистрируются экстраокципитальные спайки в передних отведениях, причем с одинаковой частотой как в унилатеральной, так и в контрлатеральной гемисфере. Редко (4% случаев) выявляются короткие генерализованные разряды медленных волн, которые могут сочетаться с фокальными спайками (15%). У 9% пациентов ЭЭГ нормальная.
Спайки могут персистировать в течение многих лет после клинической ремиссии. Они исче-
зают, когда пациент достигает пубертатного возраста [6].
ЭЭГ ночного сна характеризуется увеличением частоты затылочных спайков без изменения их морфологии и активацией патологических ЭЭГ-паттернов в фазу медленного сна [42].
В момент приступа на ЭЭГ регистрируется ритмичная дельта- или тета-активность, обычно сочетающаяся с низкоамплитудными спайками. Вначале активность унилатеральная, чаще из задних отделов, но может появляться в передних отведениях и не быть строго локализованной под одним электродом [19, 43].
Лечение
Противосудорожная терапия при затылочных эпи-лепсиях как симптоматических, так и идиопатиче-ских обычно эффективна и должна быть назначена как можно раньше [18]. Несмотря на отсутствие объективного преимущества какого-либо препарата при проведении монотерапии чаще всего предпочтение отдается карбамазепину (тегретол). Из противоэпилептических препаратов нового поколения в случаях невосприимчивости к вальпроату в последнее время успешно используется левети-рацетам (кеппра) [44].
Исключением является синдром Панайотопу-лоса. Несмотря на высокий риск развития вегетативного эпилептического статуса при синдроме Панайтопулоса, 27% пациентов имеют единственный приступ, у 47% больных отмечается от 2 до 5 приступов и только у 5% — более 10 приступов. Тем не менее активный приступный период достаточно кратковременный. У 79% пациентов через 1—2 года наступает спонтанная ремиссия без лечения. У 1/3 больных отмечается эволюция в другой вид эпилепсии, в 13% случаев — в роландическую [6]. Таким образом, длительное противосудорожное лечение пациентов с единственным или нетяжелыми эпилептическими приступами при синдроме Панайотопулоса не показано [6]. Препаратами первой очереди при лечении вегетативного эпилептического статуса являются бензодиазепины [44].
Постиктальная головная боль, возможно обусловленная серотонинергическими механизмами, купируется суматриптаном [45].
В резистентных случаях симптоматических затылочных эпилепсий рассматривается возможность нейрохирургического лечения, которое бывает эффективным у 70% пациентов с полной ремиссией приступов в 30% случаев [23, 30, 46].
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.ЛИТЕРАТУРА
1. Andermann F., Beaumanoir A, Mira E. et al. Occipital seizures and epilepsies in children. London: John Libbey & Company Ltd, 1993; 256.
2. Williamson P.D. Seizures with origin in the occipital or parietal lobes. In: P. Wolf (ed.). Epileptic seizures and syndromes, p. 383—390. London: John Libbey & Company Ltd 1994; 678.
3. Salanova V., Andermann F., Rasmussen T. et al. Parietal lobe epilepsy. Clinical manifestations and outcome in 34 patients treated between 1934 and 1988. Brain 1995; 118: 607—627.
4. Panayiotopoulos C.P. Visual phenomena and headache in occipital epilepsy: a review, systematic study and differentiation from migraine. Epilep Disord 1999; 1: 205—216.
5. Taylor I., Scheffer I.E., Berkovic S.F. Occipital epilepsies: identification of specific and newly recognized syndromes. Brain 2003;126: 753—769.
6. Panayiotopoulos C.P. Symptomatic and probably symptomatic focal epilepsies. In: C.P. Panayiotopoulos (ed.). The Epilepsies: Seizures Syndromes and Management (1st end). London: Blandon Medikal Publishing 2005; 416—429.
7. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989; 30: 389—399.
8. Engel J.Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001; 42: 796—803.
9. Hauser W.A. Incidence and prevalence of epilepsy. In: J.J. Engel, T.A. Pedley (eds.). Epilepsy: A comprehensive Textbook. Philadelphia: Lippincott-Raven Publishers 1997; 47—57.
10. Sveinbjornsdottir S., Duncan J.S. Parietal and occipital lobe epilepsy: A review. Epilepsia 1993; 34: 3: 493—521.
11. Panayiotopoulos C.P. Occipital seizures and related epileptic syndromes. In: C.P. Panayiotopoulos (ed.). Benign childhood partial seizures and related epileptic syndromes. London: John Libbey & Company Ltd 1999; 101—228.
12. Kuzniecky R., Gilliam F., Morawetz R.. et al. Occipital lobe developmental malformations and epilepsy: clinical spectrum, treatment, and outcome. Epilepsia 1997; 38: 175— 181.
13. Gobbi G., Andermann F., Naccarato S. et al. Epilepsy and other neurological disorders in coeliac disease. London: John Libbey & Company Ltd 1997; 392.
14. Gobbi G., Bertani G. Italian Working Group on Coeliac Disease and Epilepsy. Coeliac Disease and Epilepsy. In: G. Gobbi, F. Andermann, S. Naccarato, G. Banchini (eds.). Epilepsy and other neurological disorders in coeliac disease. London: John Libbey & Company Ltd 1997; 65—79.
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.15. Bercovic S.F. Progressive myoclonic epilepsies. In: J.Jr. Engel, T.A. Pedley (eds.). Epilepsy: A comprehensive textbook. Philadelphia: Lippincott-Raven Publishers 1997; 2455—2468.
16. Minassian B.A. Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediat Neurol 2001; 25: 21—29.
17. Hirano M., DiMauro S. Primary Mitochondrial Diseases. In: J.Jr. Engel, T.A. Pedley (eds.). Epilepsy. A comprehensive textbook. Philadelphia: Lippincott-Raven Publishers 1997; 2563—2570.
18. Panayiotopoulos C.P. Benign childhood partial seizures and related epileptic syndromes. London: John Libbey & Company Ltd 1999; 416.
19. Panayiotopoulos C.P. Benign childhood epilepsies with occipital paroxysms: a 15 year prospective study. Ann Neurol 1989; 26: 51—56.
20. Salanova V., Andermann F., Rasmussen T.B. Occipital lobe epilepsy. In: E. Wyllie (ed.). The treatment of epilepsy. Philadelphia: Lee & Febiger 1993; 533—540.
21. Russell W.R., Whitty C.W.M. Studies in traumatic epilepsy 3. Visual fits. Neurol Neurosurg Psychiat 1955; 18: 79— 96.
22. Penfield W, Jasper H.H. Epilepsy and the functional anatomy ofthe human brain. Boston: Little, Brown & Co 1954; 896.
23. Salanova V., Andermann F., Olivier A. et al. Occipital lobe epilepsy: electroclinical manifestations, electrocorticog-raphy, cortical stimulation and outcome in 42 patients treated between 1930 and 1991. Surgery of occipital lobe epilepsy. Brain 1992; 115: 1655—1680.
24. Babb T.L., Halgren E, Wilson C. et al. Neuronal firing patterns during the spread of an occipital lobe seizure to the temporal lobes in man. Electroencefalogr Clin Neuro-physiol 1981; 51: 104—107.
25. Blanke O., Landis T, Spinelli L., Seeck M. Out-of-body experience and autoscopy of neurological origin. Brain 2004; 127: 243—258.
26. Panayiotopoulos C.P. Elementary visual hallucination, blindness, and headache in idiopathic occipital epilepsy: differentiation from migraine. J Neurol Neurosurg Psychiat 1999; 66: 536—540.
27. Panayiotopoulos C.P. Idiopathic childhood occipital epilepsies. In: J. Roger, M. Bureau, C. Dravet et al. (eds.). Epileptic syndromes in infancy, childhood and adolescence (3rd end). London: John Libbey & Company Ltd 2002; 203—227.
28. Guerrini R.., Dravet C., Genton P. et al. Idiopathic photosensitive occipital lobe epilepsy. Epilepsia 1995; 36: 883— 891.
29. Bien C.G., Benninger F.O., Urbach H. et al. Localizing value of epileptic visual auras. Brain 2000; 123: 244—253.
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.30. Williamson P.D., Thadani V.M., Darcey T.M. et al. Occipital lobe epilepsy: clinical characteristics, seizure spread patterns, and results of surgery. Ann Neurol 1992; 31: 3—13.
31. Panayiotopoulos C.P. Differentiating occipital epilepsies from migraine with aura, acephalgic migraine and basilar migraine. In: C.P. Panayiotopoulos (ed.). Benign childhood partial seizures and related epileptic syndromes, p. 281— 302. London: John Libbey & Company Ltd 1999; 416.
32. Barry E., Sussman N.M., Bosley T.M., Harner R.N. Ictal blindness and status epilepticus amauroticus. Epilepsia 1985; 26: 577—584.
33. Williamson P.D., Engel J.Jr. Complex partial seizures. In: J.Jr. Engel, T.A. Pedley (eds.). Epilepsy: A comprehensive textbook. Philadelphia: Lippincott-Raven Publishers 1997; 557—566.
34. Ito M., Adachi N., Nakamura F. et al. Characteristics of postictal headache in patients with partial epilepsy. Ce-phalgia 2004; 24: 23—28.
35. Ito M., Adachi N., Nakamura F. et al. Multi-center study on postictal headache in patients with localization-related epilepsy. Psychiat Clin Neurosci 2003; 57: 288—290.
36. Blau J.N. Classical migraine: symptoms between visual aura and headache onset. Lancet 1992; 340: 355—356.
37. Kim S.K., Lee D.S., Kim Y.K. et al. Diagnostic performance of [18F]FDG-PET and ictal [99mTc]-HMPAO SPECT in occipital lobe epilepsy. Epilepsia 2001; 42: 1531—1540.
38. Gastaut H. Benign epilepsy of childhood with occipital paroxysms. In: J. Roger, M. Bureau, C. Dravet et al. (eds.). Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey 1992; 201—218.
39. Gastaut H. A new type of epilepsy: benign partial epilepsy of childhood with occipital spike-waves. Clin Electroen-cephalogr 1982; 13: 13—22.
40. Ferrie C.D., Beaumanoir A, Guerrini R. et al. Early-onset occipital seizure susceptibility syndrome. Epilepsia 1997; 38: 2: 285—293.
41. Covanis A., Lada C., Skiadas K. Children with rolandic spikes and ictus emeticus: Rolandic epilepsy or Panayi-otopoulos syndrome? Epilep Dis 2003; 5: 139—143.
42. Loiseau P. Benign focal epilepsies of childhood. In: E. Wyllie (ed.). Treatment of epilepsy. Principles and practice. Philadelphia: Lea and Febiger 1993; 503—512.
43. Russell W.R., Marsan C.A. Clinical ictal patterns in epileptic patients with occipital electroencephalographic foci. Neurology 1975; 25: 463—471.
44. Roger J., Bureau M., Dravet Ch. et al. Epileptic syndromes in infancy, childhood and adolescence (4edt end). London: John Libbey & Company Ltd 2005; 227—254.
iНе можете найти то, что вам нужно? Попробуйте сервис подбора литературы.45. Ogunyemi A., Adams D. Migraine-like symptoms triggered by occipital lobe seizures: response to sumatriptan. Can J Neurol Sci 1998; 25: 151—153.
46. Rasmussen T. Surgery for central, parietal and occipital epilepsy. Can J Neurol Sci 1991; 18: 611—616.
Поступила 20.08.08
cyberleninka.ru
Идиопатическая затылочная эпилепсия с ранним началом (синдром Панайотопулоса)
Е.Д. Белоусова, О.В. Гапонова Отдел психоневрологии и эпилептологии ФГУ МНИИ педиатрии и детской хирургии Росздрава, Москва
Идиопатическая затылочная эпилепсия с ранним началом (далее – синдром Панайотопулоса) представляет собой часто встречающуюся детскую возраст-зависимую предрасположенность к вегетативным приступам и вегетативному эпилептическому статусу. Для детей с этим синдромом характерно нормальное физическое и нервно-психическое развитие [1, 2].
Синдром Панайотопулоса относится к доброкачественным идиопатическим фокальным эпилепсиям, которые являются одной из самых часто обсуждаемых тем педиатрической эпилептологии [1-3]. Популярность данной проблемы обусловлена прежде всего широкой распространенностью доброкачественных идиопатических фокальных эпилепсий: педиатры, неврологи, нейрофизиологи очень часто сталкиваются с подобными пациентами. Кроме того, эти эпилептические синдромы обладают чрезвычайным клиническим и энцефалографическим своеобразием и доброкачественным исходом, который находится в некотором противоречии с драматическими клиническими проявлениями.Несмотря на достаточно четкие клинические и энцефалографические проявления, синдром Панайотопулоса был описан сравнительно недавно. Это объясняется трудностями его дифференциальной диагностики. Клинические проявления синдрома могут напоминать нейроинфекции (например, энцефалит), мигрень, гастроэнтерит и другие заболевания. Кроме того, значительная продолжительность эпилептического приступа (иногда он продолжается часами) вызывает у доктора, незнакомого с его проявлениями, сомнения в эпилептической природе клинических симптомов. Синдром был описан в результате обработки данных большого проспективного исследования, которое C.P. Panayiotopoulos начал в 1973 г. и закончил только через 30 лет [4]. У 414 из 900 наблюдавшихся им пациентов с эпилепсией отмечались афебрильные приступы в возрасте от одного до 15 лет. У 28 из этих пациентов во время приступа отмечалась рвота и вегетативные симптомы, трое из них имели повреждения центральной нервной системы, а 25 были совершенно неврологически здоровы. У двух третей из описанных 28 пациентов при энцефалографическом обследовании были обнаружены спайки в затылочных отведениях, у одной трети – межприступная энцефалограмма была либо нормальной, либо наблюдались спайки внезатылочной локализации [4].Позднее самостоятельность синдрома была подтверждена многочисленными его описаниями в различных странах у пациентов различных рас. Независимыми исследователями были описаны более 700 случаев синдрома в Азии, Европе, Японии, Канаде и Северной Америке [2].Распространенность
По данным C.P. Panayiotopoulos, синдром встречается с частотой два-три случая на тысячу общей детской популяции и составляет 6 % от афебрильных приступов у детей от года до 15 лет (или 13 % афебрильных приступов у детей в возрасте от 3 до 6 лет). Синдром одинаково часто встречается у мальчиков и девочек. C.P. Panayiotopoulos считает, что частота встречаемости синдрома может оказаться и выше, если включить в него случаи с атипичными проявлениями [1, 2, 4].Этиология и патогенез
Наиболее вероятно, что синдром Панайотопулоса является генетически детерминированным, так же как и роландическая эпилепсия. Тем не менее, семейная отягощенность по аналогичным приступам у родственников, исключая сибсов, как правило, отсутствует, хотя у них возможно наличие других форм эпилепсии [1, 2, 4]. У сибсов могут отмечаться как синдром Панайотопулоса так и роландическая эпилепсия [1, 2].Возможно, что и роландическая эпилепсия, и синдром Панайотопулоса, и другие доброкачественные идиопатические фокальные эпилепсии представляют собой проявления общих функциональных нарушений созревания коры головного мозга, которые генетически детерминированы [1, 2, 4]. C.P. Panayiotopoulos назвал эту общую генетическую основу доброкачественных фокальных эпилепсий синдромом предрасположенности к доброкачественным судорогам детства [1, 4]. Первооткрыватель синдрома считает, что у подавляющего большинства детей эта предрасположенность проявляет себя только наличием фокальных комплексов острая-медленная волна на энцефалограмме и не сопровождается клиническими проявлениями. И только у одного процента пациентов с предрасположенностью к доброкачественным судорогам детства развивается широкий спектр фокальных эпилептических синдромов – от синдрома Панайотопулоса до атипичной доброкачественной фокальной эпилепсии детства и синдрома Ландау-Клеффнера [1, 4]. Эту концепцию патогенеза разделяют и другие эпилептологи. Так, H. Doose считает, что фокальные комплексы острая-медленная волна – генетически детерминированы и наследуются аутосомно-доминантно с неполной пенентрантностью [5]. Так же как и C.P. Panayiotopoulos, H. Doose пишет о том, что только небольшой процент детей с доброкачественными фокальными разрядами дает развитие одной из идиопатических фокальных эпилепсий [1, 5].Начало клинических проявлений синдрома Панайотопулоса в определенном возрасте объясняется тем, что именно в этом периоде детства совпадает повышенная чувствительность вегетативных центров к эпилептическим разрядам и диффузная эпилептогенность коры головного мозга, которая проявляется неравномерным распределением разрядов острая-медленная волна (с их преобладанием в области затылка) при проведении электроэнцефалографии [1, 2].Клиническая характеристика
Пик начала синдрома приходится на возраст 4-5 лет, но возрастной коридор начальных проявлений значительно шире – от года до 14 лет. Тем не менее, до 76 % всех случаев начинается в возрасте от 3 до 6 лет [1].Специфические провоцирующие факторы для эпилептических приступов не описаны. Две трети всех приступов отмечаются во сне. У одного и того же пациента приступы могут быть как дневными, так и ночными. Они представляют собой необычное сочетание вегетативных симптомов (наиболее часто это тошнота, позывы к рвоте и собственно рвота), изменений поведения, девиации глаз в сторону и других более привычных проявлений эпилептического приступа [1, 2]. A. Covanis с соавт. [2] описывают следующую частоту клинических вариантов течения эпилептических приступов у пациентов с синдромом Панайотопулоса: · 10 % всех случаев – только вегетативные симптомы или вегетативный статус; · 90 % всех случаев – сочетание вегетативных симптомов и нарушения сознания; · 60-80 % всех случаев – девиация глаз; · 26 % всех случаев – гемиклонии; · 20 % всех случаев – вторично-генерализованные приступы.В Международной классификации эпилепсий и эпилептических синдромов (1989) и в предложениях по новой классификации понятие вегетативного приступа и вегетативного статуса пока отсутствует [1, 6]. H.O. Luders с соавт. [7] дали следующее определение вегетативным приступам: · вегетативные ауры представляют собой субъективное ощущение изменения активности функционирования вегетативной нервной системы; · вегетативный эпилептический приступ – эпилептический приступ либо начинается с вегетативных проявлений, либо представляет собой изолированные вегетативные эпилептические симптомы. Симптомы могут быть объективными и/или субъективными. C.P. Panayiotopoulos предлагает следующее определение вегетативного статуса – это вегетативный эпилептический приступ, который продолжается более 30 минут [1]. Далее клинические проявления синдрома рассматриваются более подробно.Вегетативные симптомы
Характерными симптомами приступа являются тошнота, позывы к рвоте и сама рвота. Классический эпилептический приступ, свойственный синдрому Панайотопулоса, выглядит следующим образом. Если приступ происходит во время бодрствования или сразу после сна, то ребенок жалуется на дурноту и плохое самочувствие. В этот момент, как правило, его тошнит, он бледен, отмечается чрезмерная потливость и гиперсаливация. Часто отмечается головная боль. Через 1-5 минут после появления первых симптомов может начаться рвота. Тошнота и позывы к рвоте заканчиваются рвотой в 74 % всех случаев. Кратность рвоты может быть различной, у части детей рвота однократна, у других детей она повторна, наблюдается в течение нескольких часов и приводит к обезвоживанию [1, 2, 4].Вместо бледности может отмечаться покраснение кожных покровов или цианоз (последнее бывает достаточно редко). Цианоз в отличие от бледности появляется не сразу, а постепенно, по мере дальнейшего развития приступа. Возможны и другие вегетативные симптомы: мидриаз или миоз, нарушения со стороны дыхательной (нерегулярное дыхание, апноэ) и сердечно-сосудистой системы (тахикардия), кашель, недержание мочи и/или кала, изменение двигательных функций кишечника. Мидриаз бывает очень выраженным, и зрачки могут перестать реагировать на свет, миоз редок и отмечается тогда, когда ребенок теряет сознание. Также возможны нарушения терморегуляции – повышение температуры как в начале приступа, так и после него [1, 2, 4].Более редкими симптомами являются гиперсаливация (6 % всех случаев) и диарея (в 3 % всех случаев).Нарушения поведения заключаются в беспокойстве, возбуждении или чувстве ужаса, которые возникают чаще всего одновременно с вегетативными проявлениями.
Девиация глаз наряду со рвотой является очень частым приступным синдромом. Отведение глаз в крайнее боковое положение может сопровождаться поворотом головы в ту же сторону и продолжаться от нескольких минут до нескольких часов. Девиация глаз может быть как постоянной, так и интермиттирующей – глаза то отводятся в сторону, то приводятся в изначальное положение.Нарушение сознания. Примерно в одной пятой всех случаев ребенок теряет сознание и обмякает, а двигательные эпилептические проявления отмечаются далеко не всегда. Данный симптом в эпилептологической литературе часто называют приступным синкопэ [1, 2]. Ребенок или внезапно, или постепенно становится дезориентированным во времени и пространстве и не реагирует на окружающее. Степень нарушения сознания может быть различной, иногда ребенок частично выполняет инструкции. Тяжесть нарушения сознания нарастает по мере развертывания симптомов приступа. Другие проявления приступа. Примерно в одной пятой всех случаев приступ заканчивается гемиконвульсиями, иногда с джексоновским маршем. В некоторых случаях наблюдается симптоматика, напоминающая приступ роландической эпилепсии – остановка речи, гемифациальный спазм и орофаринголарингеальные движения. В небольшом проценте случаев (6 %, по данным Covanis c cоавт. – 2) могут наблюдаться слепота, зрительные галлюцинации или иллюзии. Зрительные симптомы, как правило, возникают после развития вегетативной симптоматики. Характерна значительная продолжительность приступа – более чем в половине всех случаев приступ продолжается более 30 минут. Такой эпилептический приступ расценивается как статус и может продолжаться до 7 часов. Еще в половине всех случаев синдрома приступ менее продолжительный – в среднем около 9 минут. У одного и того же ребенка могут быть как продолжительные, так и короткие приступы. Пациент после приступа, даже самого продолжительного, хорошо себя чувствует, если поспит несколько часов. У него не остается никакой неврологической симптоматики. Несмотря на то что вегетативный вариант статуса при синдроме Панайотопулоса встречается довольно часто, статус гемиконвульсий или генерализованный судорожный статус встречается чрезвычайно редко.Частота приступов. Считается, что у трети пациентов (27 %) отмечается всего один приступ, у половины (47 %) – от двух до пяти приступов, и только 5 % пациентов, страдающих синдромом Панайотопулоса, имеют более 10 приступов [2]. У некоторых из этих 5 % пациентов число приступов может достигать и 50 [1, 2].
Нервно-психический статус у больных с синдромом Панайотопулоса нормальный. Интеллектуальные нарушения отсутствуют, изменений в неврологическом статусе нет.Описанные клинические проявления характерны для типичных (реже их называют классическими) случаев синдрома Панайотопулоса. Атипичные клинические проявления синдрома сам C.P. Panayiotopoulos описывает следующим образом: это эпизоды «внезапного сна или падения без судорожных проявлений», «длительные эпизоды с нарушением поведения, головной болью или другими (кроме рвоты) вегетативными проявлениям, изолированными или в комбинации друг с другом» [4]. Возникновение атипичных клинических проявлений наиболее вероятно у детей, которые имеют генетическую предрасположенность не только к фокальным идиопатическим эпилепсиям, но и к другим пароксизмальным неэпилептическим состояниям – циклическим рвотам и абдоминальной форме мигрени [2]. К атипичным случаям, по всей видимости, можно также отнести тех детей, которые наряду с типичными эпилептическими приступами имеют в силу тех или иных причин пограничные показатели IQ или какие-либо нейропсихологические нарушения [8]. Такая ситуация может сложиться у ребенка с перинатальным поражением центральной нервной системы, который одновременно страдает и синдромом Панайотопулоса.Лабораторные и функциональные методы исследованияОсновным диагностическим тестом, подтверждающим наличие синдрома, является электроэнцефалография. Изменения на электроэнцефалограмме во время приступа были документированы у 9 пациентов с синдромом Панайотопулоса [9, 10]. Типичному вегетативному приступу соответствовала унилатеральная ритмичная тета или дельта активность и отдельные спайки небольшой амплитуды. Начало эпилептической активности регистрировалось как в затылочных, так и в лобных отведениях. Клинически вегетативный статус развивался через несколько минут после начала энцефалографического эпилептического паттерна [11].
На межприступной энцефалограмме в 90 % всех случаев регистрируются комплексы острая-медленная волна, как правило, мультифокальные и большой амплитуды [1, 2, 4]. Морфология спайков такая же, как и при роландической эпилепсии. Локализация комплексов может быть самой различной (могут вовлекаться все отделы головного мозга), хотя наибольшая выраженность эпилептических изменений отмечается именно в задних отделах головного мозга. Таким образом, наиболее часто спайки наблюдаются в затылочных отведениях, чуть реже – в лобных, и еще реже – в височных [2]. В 17 % всех случаев спайки регистрируются в вертексных отведениях.Межприступные эпилептические разряды при синдроме Панайотопулоса могут быть чувствительны к определенным раздражителям. Разряды затылочной локализации могут активироваться отсутствием зрительного сосредоточения и соответственно подавляться (или становиться менее выраженными) при его наличии. В англоязычной литературе такая чувствительность получила название «fixation-off sensitivity» (от англ. прилагательного fixation-off – лишенный фиксации, закрепления, имеется в виду фиксация взора на окружающих предметах, и существительного sensitivity – чувствительность), т. е. имеется в виду особая чувствительность к зрительным раздражителям: при закрытых глазах (нет фиксации взора на предметах) регистрируется активированная эпилептическая активность, электроэнцефалограмма немедленно нормализуется после открытия глаз. Фоточувствительность или фотосенситивность (возникновение фокальных эпилептических разрядов на ЭЭГ в ответ на фотостимуляцию, иногда приводящая к развитию приступа), характерная для симптоматических затылочных эпилепсий, при синдроме Панайотопулоса встречается редко [1, 2].Межприступные эпилептические разряды любой локализации при синдроме Панайотопулоса усиливаются во время сна. Поэтому в тех случаях, когда рутинная межприступная электроэнцефалограмма бодрствования не выявляет типичных изменений, показана электроэнцефалограмма сна. В 19 % всех случаев синдрома на межприступной ЭЭГ мы можем зафиксировать своеобразную эпилептическую активность – повторные мультифокальные спайк-волновые комплексы, идентичные по морфологии (cloned-like, т. е. «клонированные» в англоязычной литературе). Такая активность может у одного и того же пациента отмечаться в разных областях головного мозга [2]. У одного и того же пациента с «клонированными» разрядами могут обнаруживаться два независимых эпилептических фокуса, расположенных унилатерально или контралатерально. Мультифокальность эпилептических проявлений при этом иногда бывает настолько выражена, что может создаться впечатление генерализованного разряда или вторичной билатеральной синхронизации. Проведение электроэнцефалографических исследований в динамике показывает, что «клонированная» мультифокальная спайк-волновая эпилептическая активность с возрастом может менять свою преимущественную локализацию у одного и того же пациента [12]. У некоторых пациентов она сначала может локализоваться в затылочных областях головного мозга, затем в области лба и затылка, а затем только в области затылка. Этот феномен получил название вторичной затылочно-лобнодолевой синхронии [12].По данным C.P. Panayiotopoulos, частота и локализация спайков не определяет клинические проявления (частоту и тяжесть приступов) и прогноз течения синдрома [1, 4]. Комплексы острая-медленная волна сохраняются на электроэнцефалограмме и после достижения клинической ремиссии. Они спонтанно исчезают в юношеском возрасте так же, как и при роландической эпилепсии. Как правило, пациентам с идиопатическими формами эпилепсии не показано нейрорадиологическое исследование. Тем не менее, если существуют малейшие сомнения в идиопатическом характере эпилепсии, показано проведение магнитно-резонансной томографии головного мозга. Примерно у 10 % пациентов с фокальными приступами, напоминающими таковые при синдроме Панайотопулоса, при нейрорадиологическом исследовании обнаруживаются структурные изменения головного мозга [2]. Как правило, это пациенты либо с симптоматической затылочной эпилепсией, либо с атипичным вариантом синдрома Панайотопулоса.Дифференциальный диагноз синдрома Панайотопулоса достаточно сложен, особенно если врач не знает о существовании эпилептического синдрома с такими необычными эпилептическими клиническими проявлениями. Диагноз особенно труден, если приступ первый, продолжается долго и ребенок находится в бессознательном состоянии.
Типичный вегетативный эпилептический приступ без нарушения сознания из-за наличия головной боли и рвоты может напоминать приступ атипичной мигрени или имитировать функциональное головокружение, которое возникает у ребенка при укачивании. Та же симптоматика, особенно если к ней добавляется диарея, может способствовать неверному диагнозу гастроэнтерита. Из-за наличия температурной реакции у врача может сложиться впечатление, что он столкнулся с фебрильными судорогами. Если у ребенка имеются нарушения сознания, то сочетание рвоты, температуры, нарушений сознания и двигательных судорожных проявлений чаще всего заставляет думать об энцефалите и менингите. Подозрение на наличие нейроинфекции неизбежно ведет к люмбальной пункции и иногда к необоснованной и дорогостоящей антибактериальной терапии. Ребенок, страдающий синдромом Панайотопулоса, действительно «трудный пациент» с точки зрения дифференциальной диагностики. В связи с этим следует подчеркнуть чрезвычайную важность электроэнцефалографического обследования. Описанные выше энцефалографические особенности синдрома настолько характерны, что в сочетании со своеобразной клинической картиной и нормальным неврологическим статусом ребенка делают диагноз синдрома Панайотопулоса практически несомненным.Для других доброкачественных идиопатических фокальных эпилепсий (роландической и идиопатической затылочной эпилепсии с поздним началом – синдром Гасто) характерны совершенно другие клинические характеристики приступов. Синдром Панайотопулоса также необходимо дифференцировать с симптоматической фокальной эпилепсией, которая может развиваться при самых разнообразных по этиологии повреждениях головного мозга. Как правило, ребенок с симптоматической фокальной эпилепсией имеет и другие варианты эпилептических приступов (не только вегетативные), а также у него отмечаются изменения неврологического статуса и/или интеллекта и/или данных магнитно-резонансной томографии головного мозга.ЛечениеБольшая продолжительность эпилептического приступа примерно в половине всех случаев и значительная выраженность вегетативных проявлений приводят к тому, что родители ребенка чрезвычайно боятся повторов приступа (им кажется, что ребенок умирает в этот момент). Поэтому родителям необходимо разъяснить особенности течения и прогноза синдрома Панайотопулоса, а также способы оказания неотложной помощи. При небольшой продолжительности течения приступа оказания неотложной помощи не требуется. Однако она необходима, если мы столкнулись с ситуацией вегетативного эпилептического статуса. Показано введение бензодиазепинов внутривенно (0,2-0,3 мг/кг в сутки) либо ректально (0,5 мг/кг в сутки) [1, 2]. К сожалению, в нашей стране не зарегистрировано ни одной ректальной формы бензодиазепинов, родители не могут воспользоваться этой лекарственной формой препарата для купирования эпилептического приступа так, как это принято во всем мире.
Как и в отношении роландической эпилепсии, профилактическая противоэпилептическая терапия, видимо, может не проводиться у отдельных пациентов ввиду того, что приступы могут быть очень редкими и длительность их существования у ребенка не очень большая (см. Прогноз) [1, 2, 13]. Тем не менее, нам кажется, что такой подход к профилактическому лечению синдрома Панайотопулоса дискутабелен. Отсутствие ректальной формы бензодиазепинов в нашей стране не дает возможности оказать ребенку неотложную помощь в момент пролонгированного приступа. Кроме того, несмотря на доброкачественность течения, у отдельных пациентов описаны такие угрожающие жизни вегетативные проявления во время приступа, как остановка сердечной и дыхательной деятельности [14]. Поэтому нам представляется, что, если мы оставляем пациента с синдромом Панайотопулоса без лечения, то мы должны быть уверены в том, что при необходимости неотложная помощь ему будет оказана в полном объеме и быстро. Если такой уверенности у нас нет, то мы должны назначить ребенку профилактическое антиэпилептическое лечение. Традиционно препаратом первой очереди выбора в лечении фокальных эпилепсий является карбамазепин [1-4]. Так как синдром Панайотопулоса редко дает атипичную эволюцию (см. Прогноз), то карбамазепин может применяться без опасения вызвать аггравацию течения эпилепсии. Большинство авторов предпочитают применение карбамазепина (Финлепсина и Финлепсина ретард) в низких или средних терапевтических дозах (10-20 мг/кг/сут) [1-14]. В отделе психоневрологии и эпилептологии наблюдаются 10 детей с синдромом Панайотопулоса, двое из них не получают лечения (у них отмечалось только по одному приступу), остальные успешно и с высокой эффективностью (у всех достигнута ремиссия) принимают карбамазепин (Финлепсин и Финлепсин ретард) в поддерживающей дозе 200 мг утром и 200-400 мг вечером либо 200-400 мг утром и 400-600 мг вечером в зависимости от возраста. Длительность лечения, за исключением отдельных случаев, не должна превышать двух лет. C.P. Panayiotopoulos рекомендует медленную (в течение 6 месяцев) отмену терапии [1]. Атипичные варианты синдрома могут оказаться резистентными к терапии [15].ПрогнозНесмотря на высокую частоту вегетативного эпилептического статуса, синдром Панайотопулоса считается прогностически благоприятным состоянием. Длительность существования эпилептических приступов небольшая и в среднем составляет один-два года [1, 2]. Тем не менее, описаны отдельные пациенты, у которых приступы отмечались и в течение 7 лет [16]. Предполагается, что риск повторного развития эпилепсии в старшем возрасте у пациента с синдромом Панайотопулоса выше общепопуляционного [1, 2]. Примерно у одной пятой всех пациентов с синдромом Панайотопулоса в старшем детском или подростковом возрасте возможно появление других приступов, наиболее часто – приступов, характерных для роландической эпилепсии (13 %). Редко, но могут наблюдаться эпилептические приступы, характерные для другой формы идиопатической затылочной эпилепсии – синдрома Гасто. Атипичная эволюция синдрома Панайотопулоса с развитием абсансов и приступов, сопровождающихся падением пациента, наблюдается чрезвычайно редко [17].
Литература 1. Panayiotopoulos C. P. A clinical Guide to Epileptic Syndromes and their Treatment. Blandon Medical Publishing; UK; 2004; 277 pp. 2. Covanis A., Ferrie C.D., Koutroumanidis M., Oguni H., Panayiotopoulos C.P. Panayiotopoulos syndrome and Gastaut type idiopathic childhood occipital epilepsy. In: Epileptic Syndromes of Infancy, Childhood and adolescence. 4th edition. Ed. Roger J. et al.; John Libbey Eurotext; 2005; P. 227-254.3. Мухин К.Ю., Петрухин А.С. Идиопатические формы эпилепсии: систематика, диагностика, терапия. М.: Арт-Бизнесс-центр; 2000; 318 стр.4. Panayiotopoulos C.P. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. London.; John Libbey; 2002.5. EEG in Childhood Epilepsy. Initial Presentation and Long-term Follow-up. Ed. H. Doose. John Libbey Eurotext; France; 2003; P.78-79.6. Engel J. Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report Of the ILAE Task Force On Classification and Terminology. Epilepsia; 2001; V.42; P. 796-803.7. Luders H.O., Noachtar S., Burgess R.C. Semiologic classification of epileptic seizures. In: Luders H.O., Noachtar S., eds. Epileptic seizures. Pathophysiology and clinical semiology. New York: Churchill Livingstone; 2000; P. 263-285.8. Oguni H., Hayashi K., Imai K. et al. Study on the early-onset variant of benign childhood epilepsy with occipital paroxysms otherwise described as early-onset benign occipital seizure susceptability syndrome. Epilepsia; 1999; V.40; P. 102-1030.9. Koutroumanidis M., Rowlinson S., Sanders S. Recurrent autonomic status epilepticus in Panayiotopoulos syndrome: videoEEG studies. Epilepsy and Behav.; 2005; V. 7(3); P. 543-547.10. Demibilek V., Dervent A. Panayiotopoulos syndrome: videoEEG illustration of a typical seizure. Epileptic Disorders; 2004; V.6; P.121-124.11. Parisi P., Ferri R., Pagani J. et al. Ictal video-polysomnography and EEG spectral analysis in a child with severe Panayiotopoulos syndrome. Epileptic Disord.; 2005; V.7(4):333-9.12. Ueno M., Oguni H., Yasuda K., Osawa M. Neurophysiological study of secondary synchronous occipito-frontopolar spikes in childhood. Clin. Neurophysiol.; 2001; V.112; P. 2106-2112.13. Ferrie C.D., Beaumanoir A., Guerrini R. et al. Early-oncet benign occipital seizure susceptability syndrome. Epilepsia; 1997; V.38; P.285-293. 14. Verrotti A., Salladini C., Trotta D. et al. Ictal cardiorespiratory arrest in Panayiotopoulos syndrome: a case report. Neurology; 2005; V.64; P.1816-1817.15. Oguni H., Hayashi K., Funtasuka M., Osawa M. Study on early-oncet benign occipital seizure susceptability syndrome. Pediatr. Neurol; 2001; V.25; P. 312-318.16. Caraballo R., Cersosimo R., Medina C., Fejerman N. Panayiotopoulos-type benign childhood occipital epilepsy: a prospective study. Neurology; 2000; V. 55; P. 1096-1100.17. Caraballo R., Astorino F., Cersosimo R. et al. Atypical evolution in childhood epilepsy with occipital paroxysms (Panayiotopoulos type); Epilept. Disord.; 2001; V.3; P.157-162.t-pacient.ru
Фокальная эпилепсия у детей: новые подходы к диагностике и лечению
Согласно определению Международной противоэпилептической лиге (International League Against Epilepsy — ILAE) 2005г., эпилептический приступ — преходящий клиническое проявление патологической избыточной или синхронной нейронной активности головного мозга. Для правильной диагностики эпилепсии сначала необходимо установить тип эпилептического припадка в соответствии с современной международной классификации эпилептических припадков, используя новое определение термина «эпилепсия».
Для правильной диагностики эпилепсии сначала необходимо установить тип эпилептического припадка в соответствии с современной международной классификации эпилептических припадков, используя новое определение термина «эпилепсия».
Первый этап диагностики — это сбор информации о самом приступе, его феноменологии, вероятности его провокации; оптимально при наличии видео самого приступа.
Второй этап диагностики — после установления факта эпилептического припадка необходимо установить его тип, согласно классификации. В 1981 г.. Принята классификация эпилептических припадков, но дискуссии по ее усовершенствованию продолжаются. В 2016 представлена обновленная рабочая классификация эпилептических припадков, которая может использоваться в практике, но будет окончательно принята позже, ожидаемо в 2017
Классификация эпилептических приступов (ILAE, 2016), базовая схема:
1. Фокальные:
- моторные;
- ходовые;
- билатеральные тонико-клонические.
2. Генерализованные:
3. С неизвестным началом:
4. Неклассифицированные.
Для всех припадков необходимо указать уровень нарушений сознания: припадок без нарушения сознания, с нарушением сознания, с неизвестной сознанием.
Классификация фокальных эпилептических приступов (ILAE, 2016):
1. Моторные:
- тонические;
- атонические;
- миоклонические;
- клонические;
- эпилептические спазмы;
- гипермоторных.
2. ходовые:
- сенсорные;
- когнитивные (галлюцинации, дежавю, иллюзии, нарушение внимания, афазия, навязчивые мысли) эмоциональные (ажитация, агрессия, плаксивость, смех)
- вегетативные (бради-, тахикардия, асистолия, ощущение холода или жара, покраснение или бледность кожи, гастроинтестинальные нарушения, лихорадка, гипер-, гиповентиляция, тошнота, рвота, пилоэрекция и др.)
- автоматизмы (агрессии, мануальные (в руках), орофациальная, сексуальные, вокализация, сложные движения в виде ходьбы или бега, раздевание).
3. Билатеральные тонико-клонические (в прошлой классификации — со вторичной генерализацией).
Для фокальных приступов также для каждого типа приступа необходимо отметить степень сознания: нападение без нарушения сознания, с нарушением сознания, с неизвестной сознанием.
В 2016 ILAE внесла некоторые изменения в терминологию приступов. Так, рекомендуется заменить термин «частичные» нападения на «фокальные» (с / без нарушения сознания и с неизвестной сознанием), «комплексные парциальные» нападения — на «фокальные с нарушенным сознанием».
Почти 60% эпилептических припадков является локальными и только 23% — генерализованными тонико-клоническими. Согласно данным современных исследований, эпилепсия является системным заболеванием головного мозга, связанным с нарушением нейрональных связей, а не только локальным нарушением функции головного мозга. По привлечению многих нейрональных связей эпилептические припадки могут возникать из неокортикальных, таламо-кортикальных, лимбических и стволовых отделов.
Третий этап диагностики. Помимо установления типа нападения, необходимо провести топической диагностике нападения, то есть установить местонахождение эпилептического очага, если эти нападения являются локальными. Приступы, возникающие вследствие чрезмерного патологического возбуждения определенной группы нейронов в различных долях головного мозга, имеют свои характеристики.
Темпоральные эпилептические припадки: дневные, возникают с частотой несколько раз в месяц, редко осложняются эпилептическим статусом, манифестируют ходовые феноменами — часто аурой (вегетативной, психотической, часто с нарушениями сознания, автоматизмами (оральными, вербальными), моторные феномены жидкие, из них могут быть дистонические установки, постиктальном нарушения сознания.
Лобные эпилептические припадки имеют следующие характеристики: частые кластерные атаки, кратковременный течение (20-40 с), часто развитие во сне, часто с вторичной генерализацией к эпистатусного течения, полиморфные ауры с резким началом, рано дебютируют передоминантные моторные изменения — парезы, параличи, дисграфия и т.п., могут протекать с нарушением сознания, восстановление после нападения быстрое. Чаще всего диагностируют тонические лобные нападения (около 64%), затем клонические (36%) и эпилептические спазмы (36%).
Для фокальных эпилептических припадков с очагами в задней коре характерны зрительные, соматосенсорные, вегетативные, вкусовые ауры, адверсивни нападения и опсоклонусы глаз, моргание, анозогнозия, акалькулия, апраксия, алексия.
Четвертый этап диагностики. Типы эпилептических припадков является основой для установления формы эпилепсии, согласно классификации 1989 Согласно определению 2005г., Эпилепсия — расстройство головного мозга, характеризующееся устойчивой склонностью к эпилептическим припадкам, а также Нейробиологические, конитивнимы, психологическими и социальными последствиями этого состояния. Это определение эпилепсии предусматривает развитие как минимум одного эпилептического приступа. Термин «расстройство» недостаточно понятный для пациентов и призменшуе серьезность состояния, поэтому ILAE и Международное бюро по изучению эпилепсии (International Bureau for Epilepsy — IBE) недавно приняли совместное решение считать эпилепсию болезнью. В 2014 принято новое практическое определение эпилепсии, согласно которому эпилепсия — заболевание головного мозга, которое отвечает следующим состояниям:
- не менее двух неспровоцированных или рефлекторных эпилептических припадков с интервалом не менее 24 ч;
- один неспровоцированный (рефлекторный) эпилептический припадок и вероятность повторных приступов, которая соответствует общему риска рецидива (> 60%) после двух неспровоцированных эпилептических припадков в следующие 10 лет
- диагноз эпилептического синдрома (например синдром Веста).
Критерии «завершение» эпилепсии включают достижение определенного возраста у пациентов с формой эпилепсии, зависит от возраста, или отсутствие эпилептических приступов в течение 10 лет у пациентов, не получавших противосудорожных препаратов в течение более 5 лет. Рабочая группа работала над термином «излечения», который указывает на то, что риск эпилептических припадков не выше, чем у здоровых людей, однако у пациентов с эпилепсией в анамнезе такой низкий риск никогда не достигается. Термин «ремиссия» недостаточно понятен и не говорит об отсутствии болезни. Рабочая группа предложила термин «завершения» эпилепсии, который свидетельствует, что эпилепсии у пациента уже нет, однако нельзя с уверенностью исключить появление приступов в будущем. Риск рецидива приступов зависит от формы эпилепсии, возраста, этиологии, лечения и других факторов. Например, при ювенильной миоклонические эпилепсии риск повторных приступов остается высоким в течение десятилетий. Структурные поражения головного мозга, врожденные пороки сопровождаются постоянной склонностью к эпилептическим припадкам. При исследовании 347 детей с отсутствием приступов в течение не менее 5 лет (без приема противосудорожных препаратов) поздние рецидивы зарегистрированы у 6% детей.
Причины эпилепсии
Более половины детей с эпилепсией страдают идиопатической формы заболевания, при которой нет других установленных причин, чем генетические. В классификации AT Berg и соавторов (2010) вместо термина «идиопатическая» предложено «генетическая», то есть в результате уже известных и предполагаемых генов. Многие гены уже известно (аутосомно-доминантная ночная лобная эпилепсия и др.).
Эпилепсия, причина которой известна и она не связана с генетическими факторами, называется симптоматической (структурной / метаболической) , по терминологии AT Berg и соавторов (2010). В этом случае эпилепсия является вторичным результатом конкретных установленных структурных или метаболических заболеваний:
- повреждения вещества головного мозга вследствие хронической гипоксии и асфиксии в родах, родовая травма, субдуральные гематомы, перенесенные врожденные TORCH-инфекции;
- метаболические заболевания (нарушение обмена аминокислот, углеводов и др.), которые сопровождаются полиорганной симптоматикой, кроме эпилепсии; митохондриальные болезни;
- врожденные пороки развития головного мозга;
- хромосомные синдромы: синдромы Ангельмана, Дауна, ломкой Х-хромосомы и др .;
- наследственные невротические синдромы (Факоматозы): туберозный склероз и др .;
- черепно-мозговая травма;
- сосудистые артериовенозные мальформации головного мозга;
- перенесенный инсульт.
Существует еще одна группа эпилепсии с неизвестной этиологией (ранее называлась криптогенная эпилепсия), причина приступов при которой пока не установлена, она может быть генетической или структурно-метаболической.
Прогноз зависит от объема и причины поражения мозга. Так, тяжелые пренатальные поражения могут трудно поддаваться лечению.
Международная классификация эпилепсии и эпилептических синдромов ILAE 1989 (сокращенный вариант)
Локализационно обусловленные (фокальные, парциальные) эпилепсии и синдромы
1. Идиопатические (генетические):
- доброкачественная эпилепсия детского возраста с центрально-темпоральными спайками на электроэнцефалограмме (ЭЭГ) (роландическая)
- доброкачественная детская эпилепсия с затылочными приступами (синдром Гасто)
- доброкачественная парциальная затылочная эпилепсия с ранним дебютом (синдром Панайотопулоса) первичная эпилепсия при чтении;
- аутосомно-доминантная лобная эпилепсия.
2. Симптоматические (структурные / метаболические):
- хроническая прогрессирующая парциальная эпилепсия детского возраста (Кожевникова) синдром Расмуссена;
- эпилепсия, которая характеризуется приступами, которые вызываются специфическими провоцирующими факторами;
- височная эпилепсия;
- лобная эпилепсия;
- теменная эпилепсия;
- затылочная эпилепсия.
3. Криптогенные (неизвестные).
1. Идиопатические (генетические) эпилепсии
Доброкачественная детская эпилепсия с центро-темпоральными спайками на ЭЭГ (роландическая эпилепсия)
Частота в популяции 21 на 100 тыс. Детского населения.
Диагностируют у 15-25% среди всех детей школьного возраста с эпилепсией. Болезнь дебютирует в возрасте 4-10 лет с максимумом в 9 лет. Мальчики болеют чаще, чем девочки. Клинически проявляется характерными признаками: начало с сенсомоторной ауры, появляются «горловые» звуки или анартрия, гемифациальный короткие моторные припадки ночью при засыпании и просыпание, в 20% — также фациобрахиальни судороги, в 25% случаев в дебюте наблюдают вторично-генерализованные припадки. Продолжительность приступов: простые — 30-60 с, вторично-генерализованные — до 1-2 мин с частотой приступов 2-6 раз в год (в возрасте до 6 лет в дебюте болезни — частые припадки). Эта форма является доброкачественной, то есть кроме эпилептических припадков нет изменений в неврологическом статусе, когнитивной сфере — ребенок может учиться в средней общеобразовательной школе. Болезнь имеет доброкачественное течение; ремиссия наступает обычно в 98% пациентов до достижения пубертатного периода.
Эпилептиформные изменения между приступами в 90% случаев;
типично: доброкачественные эпилептиформные изменения детства (ДЕЗД) в центрально-височных отведениях (по типу QRST на ЭКГ), но в возрасте 3-5 лет — в задньоскронево-затылочных отведениях; у 30% детей регистрируют только ночные ЭЭГ-феномены (во время медленного сна — пик-волновые комплексы) нормализация ЭЭГ происходит значительно позже, чем клиническая ремиссия.
В лечении применяют только монотерапии одним из препаратов первой линии препараты вальпроевой кислоты, карбамазепин, ламотригин, окскарбазепин, габапентин, топирамат, леветирацетам. Но есть данные о возможной вторичной билатеральной синхронизации, особенно при применении карбамазепина и окскарбазепина.
Доброкачественная парциальная затылочная эпилепсия с ранним дебютом (синдром Панайотопулоса)
Приступы возникают редко (до 5-7 при жизни), преимущественно во время сна, проявляются девиацией глаз в сторону, нарушением сознания по типу дезориентации, активным рвотой, после этого возникает приступообразная головная боль. У половины детей приступы могут быть длительными — в течение нескольких часов с потерей сознания (обмороки), сопровождаются рвотой, девиацией глаз, клоническими гемисудомамы, постиктальной головной болью.
Детская затылочная эпилепсия с поздним дебютом (синдром Гасто)
Приступы регистрируют чаще, чем при синдроме Панайотопулоса (1 раз в неделю — 1 раз в месяц). Болезнь начинается в возрасте 3-15 лет, с максимумом в 8 лет. Клиническое ядро - простые парциальные сенсорные нападения — зрительные галлюцинации в периферическом поле зрения, гемианоптични галлюцинации, иллюзии с ощущением боли в глазах, моргание, поворот глаз и головы в противоположную от эпилептогенного очага сторону. Длительность приступов составляет секунды-минуты. В конце приступа характерны жалобы на сильную головную боль с рвотой (у 50% больных). Может быть вторичная генерализация с тонико-клоническими судорогами. При синдромах Панайотопулоса и Гасто изменений при оценке неврологического статуса и когнитивной сферы ребенка нет.
- ДЕЗД в затылочных отведениях у 90% больных между приступами;
- основной фон без изменений;
- У 30% детей могут быть изменения в височных отведениях;
- типично: исчезновение патологического паттерна при открывании глаз высокая фотосенситивнисть;
- ночной ЭЭГ-видеомониторинг: в стадии медленного сна — нарастание ДЕЗД-комплексов (ранняя диагностика болезни) нормализация ЭЭГ-картины до достижения возраста 15 лет.
В лечении применяют принцип МОНОТЕРАПИИ одним из следующих препаратов — карбамазепин, препараты вальпроевой кислоты, окскарбазепин, топирамат, ламотригин.
Указанные формы эпилепсии также считают доброкачественными. Полная ремиссия при синдроме Панайотопулоса возникает до достижения возраста 9 лет, при синдроме Гасто — 15 лет.
Аутосомно-доминантная лобная эпилепсия
Гены CHRNA4 , CHRNA2 , CHRNB2 локализованы в локусах 20q13, 8q, 1p21 соответственно. Эта форма идиопатической эпилепсии начинается чаще в возрасте 7-12 лет. Характерны ночные приступы (после засыпания, за 2-3 часа до просыпание). Начало возникает с вокализации (обычно — крик), при этом глаза открыты. По характеру нападения простые и сложные парциальные.
Характерен полиморфизм клиники нападений — сложные двигательные акты: ребенок садится, чешет нос, голову, делает гримасы, жевательные движения, становится на четвереньки, раскачивается, делает педалювальни или боксувальни движения. В 70% случаев может быть аура (неприятные звуки, генерализованный озноб, головокружение) — ребенок просыпается. Продолжительность приступа — до 1 мин. За ночь может быть несколько нападений. При этой форме эпилепсии существует тенденция к серийности и «светлый промежуток» (отсутствие приступов в течение 2-3 мес). Обследование не выявляет изменений в неврологическом статусе, интеллекте и речи.
Характеристики ЭЭГ:
- основной фон — без изменений;
- в состоянии вне сном — без эпилептических феноменов;
- основная диагностическая методика — ночной ЭЭГ-видеомониторинг, во время которого регистрируют региональную активнисть в лобных, лобно-височных отведениях.
Лечение сложное, чаще эффективная политерапия: карбамазепин, препараты вальпроевой кислоты, топирамат, ламотригин, леветирацетам или комбинация базовых препаратов.
Эта форма эпилепсии требует проведения дифференциального диагноза с симптоматической лобной эпилепсией, при которой на ЭЭГ наблюдается замедление основного ритма, неврологический статус — без очаговых изменений, при нейровизуализации — органические изменения вещества мозга. Также следует провести дифференциальный диагноз с парасомнии, при которых на ЭЭГ отсутствуют эпилептические паттерны.
2. Симптоматическая (структурная / метаболическая) эпилепсия
Лобная эпилепсия
Среди всех симптоматических и вероятно симптоматические (криптогенным) эпилепсии симптоматическая лобная эпилепсия составляет 20%. Она может начинаться в любом возрасте в зависимости от причины. В зависимости от локализации эпилептогенного очага выделяют 7 форм лобной эпилепсии, и каждая проявляется своими типами приступов. В целом характеризуется локальными простыми или сложными приступами, которые возникают в лобной коре — контралатеральные клонические судороги, одно-, двусторонние тонические судороги, которые заканчиваются параличом Тодда, сложные автоматизмы, которые выглядят как молотильные движения конечностями, раскачивания туловища, педалювальни движения ногами. Эпилептические разряды в дополнительной лобной моторной области проявляются сложными фокальными приступами в виде тонических судорог рук, классической «позы фехтовальщика», адверсия головы, двустороннего разгибание туловища, шеи, вокализации. Активность в области поворота головы и глаз проявляется адверсия глаз в противоположную сторону, морганием. Сознание сохранено или теряется не полностью. Приступы с фокусом в центральной зоне (участок коры у роландова борозды) характеризуются джексоновского маршем или строго локализованными клоническими или тоническими судорогами, судорогами лица, потерей мышечного тонуса. При раздражении кожи может возникнуть моторный нападение без нарушения сознания, фациальные судороги с глотательными, жевательными движениями, саливацией с чувством другого вкуса, гортанные симптомы. Приступы ночные, очень часто, краткосрочные.
В неврологическом статусе выявляют парезы, атаксия, интеллектуальные и речевые нарушения.
Характеристики ЭЭГ:
- основная фоновая активность замедлена;
- региональная епиактивнисть (острые волны, комплексы острая — медленная волна, пик-волны) бифронтальна или диффузная активность;
- вторичная билатеральная синхронизация (признак ухудшения болезни, появление когнитивных нарушений).
Лечение сложное. Очень часто приступы резистентные к адекватной терапии. Необходимо начинать с монотерапии препаратами первой линии в адекватной дозе, а дальше переходить на комбинацию препаратов с различными механизмами действия, согласно Единообразным протоколом лечения эпилепсии у детей 2014 Препараты первой линии — карбамазепин (при вторичной билатеральной синхронизации противопоказан), окскарбазепин, топирамат, второй — препараты вальпроевой кислоты, ламотригин, третьей — комбинации препаратов.
Височная эпилепсия
Частая форма всех симптоматических эпилепсии (30-35%). Дебют отмечают в разном возрасте (чаще школьном). Частые причины: последствия гипоксически-ишемической энцефалопатии в виде глиозом, врожденные пороки развития (кортикальная дисплазия), арахноидальные кисты, последствия перенесенного энцефалита, формирования склероза гиппокампа. Приступы могут быть у одного пациента с / без нарушения сознания. Приступы длительные — 1-2 мин. Вегетативные проявления, психические и сенсорные симптомы имеющиеся в течение всего нападения или только в начале в виде ауры, дальше продолжается фокальный нападение с нарушением сознания с билатеральных тонико-клоническими судорогами. Выделяют две формы височной эпилепсии в зависимости от эпилептогенного очага: медиальная (амигдалу-гипокампальна) и латеральная (неокортикальных) эпилепсия.
Медиальная (амигдалу-гипокампальна) эпилепсия занимает 65% всех височных эпилепсий и обусловлена наличием фокуса в медиальных отделах височной доли. Причина — гипокампальни атрофии, часто у пациентов, у которых в возрасте до 3 лет наблюдались комплексные фебрильные судороги, особенно длительные односторонние атаки (в 40% случаев). После 5-6-летнего периода ремиссии начинаются фокальные частые резистентные нападения, то есть развивается хроническая эпилепсия.
Клиническая основа этого подтипа эпилепсии:
- фокальные приступы без нарушения сознания — изолированная аура (вегетовисцеральных, обонятельные и вкусовые галлюцинации), психические феномены — состояние сна, деперсонализации, дереализации, страх, аффект, радости, ороалиментарни автоматизмы с сохраненной сознанием, дистонического положения контралатеральной руки, в ипсилатерально руке могут быть простые автоматизмы;
- фокальные припадки с изолированным выключением сознания и автоматизмами без судорог (диалептични припадки).
Для латеральной (неокортикальной) эпилепсии характерны:
- слуховые галлюцинации
- зрительные яркие галлюцинации (панорамные виды)
- вегетативные приступы (несистемного головокружения, «височные синкопе» — медленное падение без судом с дистонического установкой конечностей, автоматизмами)
- пароксизмальная сенсорная афазия.
Кроме частых приступов, при грубых очаговых изменениях вещества мозга дети имеют неврологический дефицит контралатерально очагу (парез), эмоциональные и интеллектуальные нарушения.
Характеристики ЭЭГ:
- у 50% больных нормальная картина ЭЭГ между приступами; необходимый стандарт исследования — инвазивные электроды;
- у 30% больных наблюдаются епипатерны между приступами;
- при медиальной эпилепсии — изменения в передньоскроневих отведениях;
- ЭЭГ-очаги могут не совпадать с морфологическим очагом на магнитно-резонансной томографии (МРТ) — формирование «зеркального» очага;
- характерный ЭЭГ-феномен в дебюте — продолжено региональное замедление активности;
- провокация — иногда депривация сна;
- ночная ЭЭГ свидетельствует о 65% изменений между приступами.
На интериктальном ЭЭГ — передний височная фокус спаек, пароксизмальный тета-ритм.
Характеристики изменений на МРТ головного мозга при медиальной эпилепсии — атрофия гиппокампа, усиленный сигнал на Т 2 от гиппокампа. Склероз гиппокампа прогрессирует.
Лечение хирургическое. Прогноз после хирургического лечения хороший. Медикаментозное лечение сложное и не всегда эффективное; часто применяют политерапию.
Теменная эпилепсия
Приступы субъективные, поэтому обнаружить их сложно, особенно у детей младшего возраста. Характерные соматосенсорные нападения в виде чувствительного джексоновского марша, часто связанные с моторными феноменами. Соматосенсорные симптомы могут быть положительными и отрицательными, возможны боли в животе, тошнота, иллюзия движения, отсутствие чувства тела (асоматогнозия), головокружение, дезориентация в пространстве. Нарушение восприятия и речи (с привлечением доминантного полушария), постуральные или ротаторные движения, при распространении импульса могут развиваться зрительные симптомы (затылочно-височно-теменная), контралатеральные или ипсилатерально движения с дистонического положением конечности в противоположную сторону или в сторону вовлеченной полушария . Зрительные иллюзии (макропсия, микропсия, метаморфопсии) свидетельствуют о наличии разрядов в задних отделах теменной коры и париетотемпороскроневои доли.
Затылочная эпилепсия
Регистрируют у 5% детей всех симптоматических и криптогенным форм.
Эпилептические разряды по первичной зрительной коры проявляются:
- глазодвигательными расстройствами (нистагм, девиация глаз в противоположную сторону, двусторонний миоз)
- локальными приступами без нарушения сознания в виде зрительных галлюцинаций, иллюзий, пароксизмальной амавроза, сужение полей зрения;
- локальными приступами с нарушением сознания и с билатеральных тонико-клоническими припадками;
- вегетативные расстройства конце приступа (головная боль, рвота)
- акалькулия, апраксия.
- Неврологический дефицит зависит от причины эпилепсии. Часто обнаруживают глазодвигательные нарушения (нарушение конвергенции, косоглазие).
Характеристики ЭЭГ:
- между приступами может быть в норме;
- замедление основного фона;
- одностороннее подавление альфа-ритма при грубых органических изменениях;
- ЭЭГ-паттерны не изменяются при открывании глаз (дифференциальный диагноз с идиопатической затылочной эпилепсией)
- распространение эпиактивности на височные отведения;
- провокация — фотостимуляция.
Злокачественные мигрирующие фокальные приступы раннего детства (синдром Коппола — Дюлак)
Относительно новая форма фокальной эпилепсии.
характеристика:
- этиология неизвестна (вероятно генетического происхождения);
- начало возраст 6 мес;
- нормальное развитие к дебюту;
- моторный и интеллектуальный регресс;
Приступы:
- фокальные моторные;
- билатеральные тонико-клонические;
- вегетативные (апноэ, цианоз)
- в виде серий и кластеров (2-5 суток), короткие ремиссии.
прогрессирующая микроцефалия; на ЭЭГ — типичный фокальный паттерн в различных отведениях;
на МРТ — норма.
medictionary.ru